放射性药品的放化纯度分析方法概述
2021-06-14张云,杨柳,姜华
张 云,杨 柳,姜 华
(原子高科股份有限公司,北京 102413)
放射性药品是指用于临床诊断或者治疗的放射性核素制剂或者其标记化合物[1]。放化纯度是评价放射性药品质量的关键质控项目之一,任何放射性药品均可能存在影响放化纯度的放射性化学杂质,其主要来源是:(1) 制备过程的副反应或者中间体纯化不完全;(2) 放射性核素与制剂中辅料成分的反应;(3) 储存期间射线辐射引起活性成分分解、活性成分化学不稳定(水解、氧化-还原等)等。这些放射化学杂质可能会影响药品在人体的生物分布,干扰诊断准确性或者引起治疗照射不足,并对人体其他组织器官造成不必要的辐射损伤等。因而必须严格控制放化纯度,保证药品的安全性和有效性[2-3]。本文通过查阅《中国药典》2020年版(ChP 2020)、《欧洲药典》10.2版(EP 10.2)和《美国药典》43-NF38版(USP 43-NF38)收录有关放化纯度测定的相关指导原则[1,4-5]、各国放射性药品质量标准以及相关文献资料,对放化纯度测定常用分析方法的特点、分离原理和应用进行总结概述,为建立更科学、合理的放射性药物质量标准提供参考。
1 指导原则及测定方法
《中国药典》2020年版(ChP 2020)、《欧洲药典》10.2版(EP 10.2)和《美国药典》43-NF38版(USP 43-NF38)收录的放射性检定法相关指导原则对放化纯度测定均作了规定,相关的指导原则以及测定方法比对列于表1。放化纯度的测定过程一般分为两个阶段,首先通过分离技术将不同的化学物质进行分离,再用合适的放射性测量仪器测定每种化学物质的放射性活度。在ChP 2020通则1401放射性药品检定法中收录的放射性化学测定方法主要包括薄层色谱法(thin-layer chromatography, TLC)、纸色谱法(paper chromatography, PC)和电泳法(electrophoresis, EP),经这些方法分离后所获得的薄层板/色层纸置于放射性薄层扫描仪或者γ计数器中测得放射性分布,从而得到放化纯度;另外,也规定了经过验证并且能有效分离各种放射化学杂质的其他分离分析方法,如高效液相色谱法(high performance liquid chromatography, HPLC)、柱色谱法(column chromatography, CC)等可用于放化纯度测定,但ChP 2020收录的各放射性药品质量标准放化纯度测定项下除薄层色谱法和纸色谱法以外未收录其他方法。而EP 10.2和USP 43-NF38中相关通则的规定与品种标准相结合,除TLC、PC、EP这些常规方法之外,还收录了液相色谱仪串联紫外或电化学等常规检测器与放射性流量检测器测定放化纯度的分析技术,即HPLC法。HPLC法适用于不同类型的样品,分离效能、检测灵敏度高,尤其是对于结构比较复杂的标记化合物,采用HPLC法或与其他方法联合的分离检测技术手段,可较全面地对放射性药品的杂质谱进行分析与控制。

表1 ChP 2020、 EP 10.2 、 USP 43-NF38相关通则中放化纯度测定方法比较Table 1 Comparison of radiochemistry purity measurement methods in the general monograph of ChP 2020, EP 10.2 and USP 43-NF38
2 主要分析方法
2.1 薄层色谱法
薄层色谱法又称薄层层析法,是将固定相均匀地涂铺在具有光洁表面的玻璃、塑料或金属板上形成薄层,在此薄层上进行色谱分离的一种平面色谱方法[6]。薄层色谱法具有操作简便、成本低、适用范围广等优点,但是对复杂样品的分离效率以及鉴别能力有待提高。
其分离原理包括极性吸附和液-液分配。(1) 极性吸附,一般以吸附剂为固定相的薄层色谱法,指吸附剂对各组分具有不同的吸附能力,展开剂对吸附于其中各组分的溶解、解吸附能力不相同,组分在随流动相迁移过程中不断被吸附、解吸附、再吸附、再解吸;(2) 液-液分配,一般以液体为固定相的薄层色谱法,主要是利用各组分在固定相与流动相之间的分配系数不同,分配系数大的组分在板上的迁移速度慢,反之亦然,从而在薄层板上产生差速迁移而分离[6]。
按照固定相种类,薄层板可分为正相薄层板(硅胶薄层板、聚酰胺薄层板等)和反相薄层板(如C18键合相薄层板等)[7]。
2.1.1正相薄层色谱法 正相薄层板主要以硅胶为主,适用于大部分化学结构的分离。硅胶薄层板作为固定相,展开剂为单组分溶剂时,分离过程相对简单。以锝[99mTc]双半胱乙酯注射液为例,分子结构示于图1。EP 10.2与USP 43-NF38收录的放化纯度检查方法均采用快速硅胶板作为固定相和乙酸乙酯作为展开剂,属于典型的正相薄层色谱法,通过吸附分离原理实现活性成分锝[99mTc]双半胱乙酯与锝[99mTc]双半胱乙酯的酯基水解物等六种杂质的分离[4-5]。
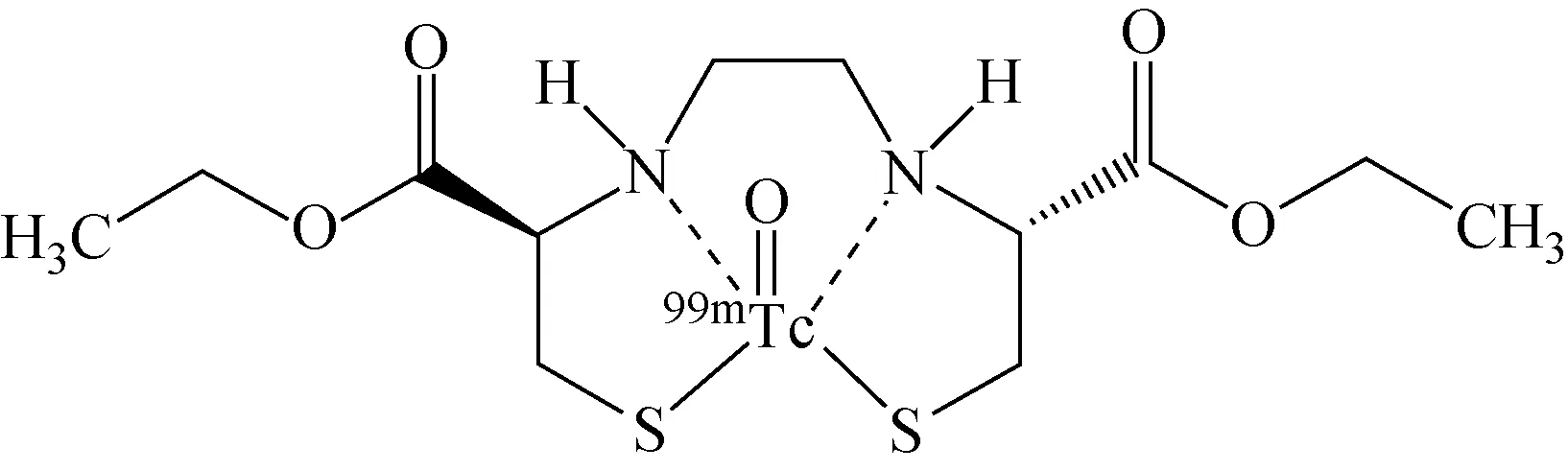
图1 锝[99mTc]双半胱乙酯的分子结构[5]Fig.1 The molecular structure of 99mTc-ECD[5]
以二元、三元甚至多元溶剂组成的展开剂体系占多数,在混合展开剂中占比较大的主要溶剂起调整改善分离物质的比移值(retardation factor,Rf)及对某些物质的选择作用,一般主要溶剂选用不易形成氢键的溶剂,或极性比分离物质低的溶剂,否则将使被分离物质的Rf太大甚至跟随溶剂前缘移动,不易将各组分分离[8]。值得注意的是,国内外药典中收录的多种锝标记药物,如锝[99mTc]喷替酸盐注射液、锝[99mTc]亚甲基二膦酸盐注射液、锝[99mTc]焦磷酸盐注射液、锝[99mTc]葡庚糖酸盐注射液等,以快速硅胶板作为固定相,以水相和有机相作为双体系展开剂,正是利用了活性成分在不同展开剂体系下吸附作用效果不同来实现主峰锝[99mTc]标记化合物与杂质胶体锝[99mTc]、高锝[99mTc]酸盐的分离[4-5, 9]。
临床应用于肾及泌尿系统功能检查的邻碘[131I]马尿酸注射液,分子结构示于图2,在EP 10.2收录的质量标准放化纯度测定项下采用GF254硅胶作为固定相,水-冰醋酸-丁醇-甲苯(体积比为1∶4∶20∶80)混合体系作为流动相,甲苯作为主要溶剂起着调整改善主峰邻碘[131I]马尿酸盐与杂质峰碘[131I]离子、邻碘[131I]苯甲酸的分离作用,对于各放射性成分的鉴别,采用了非放射性物质碘离子、邻碘马尿酸和邻碘苯甲酸参照溶液在紫外波长254 nm下进行定位,并与放射性分布进行对比,得到碘离子、邻碘马尿酸和邻碘苯甲酸的Rf分别位于薄层板的原点、中部和前沿,分离效果较好[4]。
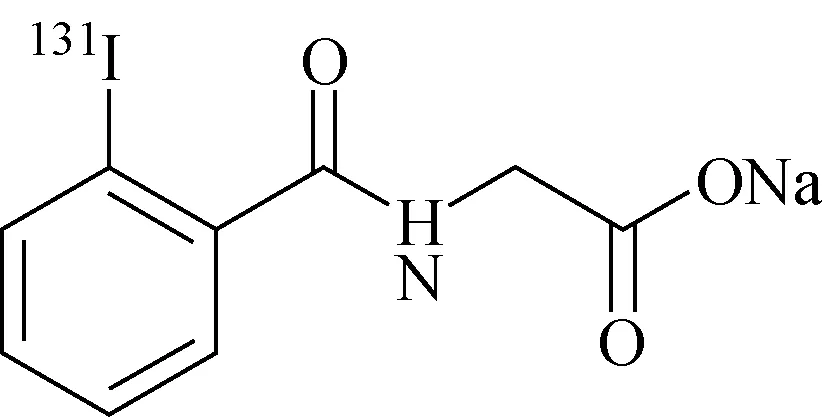
图2 邻碘[131I]马尿酸的分子结构[4]Fig.2 The molecular structure of sodium iodohippurate [131I][4]
以聚酰胺为载体的薄层板,由于聚酰胺分子内存在很多酰胺基,可与酚类、酸类、醌类及硝基化合物等形成氢键,因而对这些物质产生了吸附作用[8]。一般在水中形成氢键的能力最强,在有机溶剂中形成氢键的能力较弱,在碱性溶剂中形成氢键的能力最弱。ChP 2020锝[99mTc]甲氧异腈注射液和锝[99mTc]双半胱乙酯注射液采用聚酰胺-6作为固定相。锝[99mTc]双半胱乙酯注射液放化纯度测定所用固定相和展开剂为聚酰胺-6薄片和甲醇-二氯甲烷-水(体积比为80∶15∶5),在该展开体系下,主峰的Rf为0.9,杂质峰高锝[99mTc]酸盐和胶体锝[99mTc]的Rf在原点附近[9]。
2.1.2反相薄层色谱法 反相薄层色谱法是以反相硅胶薄层板作为固定相的一种薄层色谱法,分离机制属于分配色谱,色谱柱主要为C2、C8和C18等烷基链修饰的硅胶,可用于分析非极性化合物(类酯、芳香族化合物)和极性化合物(碱性和酸性化合物)[6]。
以氟[18F]多巴为例,作为L-多巴的类似物,结构示于图3a,在临床上是研究大脑突触前多巴胺能神经功能的正电子显像剂,可用于早期帕金森氏病的诊断[10]以及肿瘤的鉴别诊断研究[11],《欧洲药典》收录其放化纯度分析方法采用TLC十八烷基硅烷基手性硅胶板为固定相和甲醇-水(体积比为50∶50)为展开剂的反相薄层色谱法,从而有效地将活性成分左旋氟[18F]多巴和氟[18F]离子、活性成分的光学异构体右旋氟[18F]多巴(结构如图3b所示)这两个杂质峰进行分离,Rf值分别为0.3、0.5和0,并通过茚三酮与氨基酸或肽的特征反应对活性成分与其光学异构体进行区分鉴别。
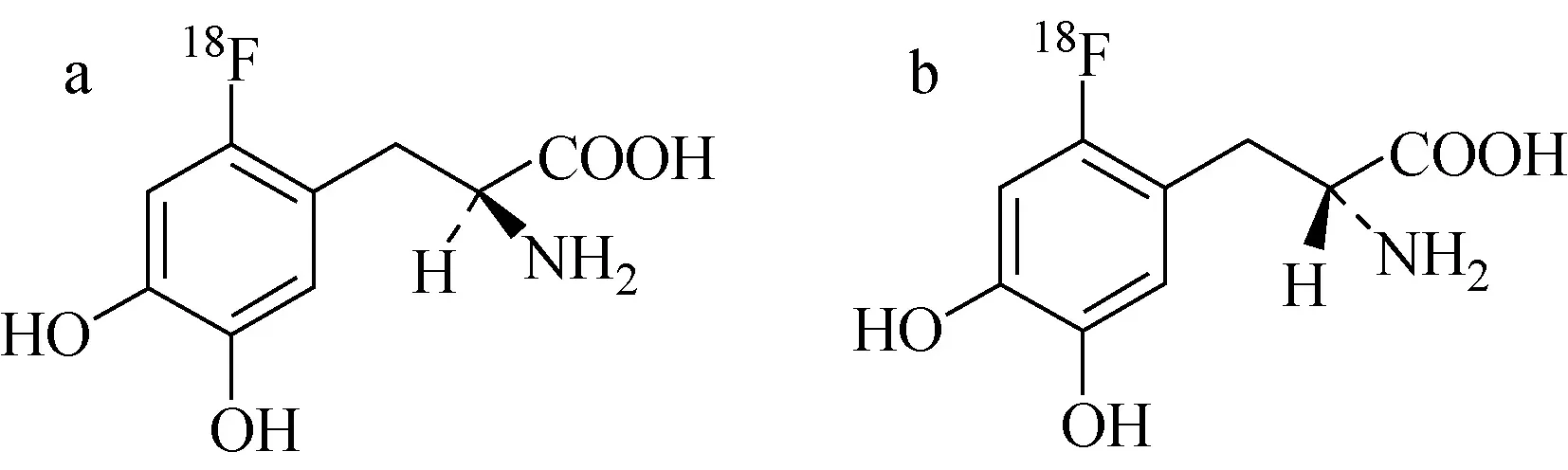
图3 左旋氟[18F]多巴(a)和右旋氟[18F]多巴(b)的分子结构[4]Fig.3 The molecular structure of L-[18F]Fluorodopa (a) and D-[18F]Fluorodopa (b)[4]
2.2 纸色谱法
纸色谱法是以纸为载体,以纸上所含水分或其他物质为固定相,用展开剂展开的分配色谱法[7]。主要分为正相纸色谱法和反相纸色谱法[6]。纸色谱法具有操作简便、成本低等优点,对于复杂样品的分离效率及鉴别能力有待提高。
其分离原理如下:纸的材质是纤维素,分子结构中含有许多羟基,有较强的亲水性,通常能吸收约20%的水分,这些水作为分配色谱中的固定相,或低极性溶剂处理纸后作为固定相,当展开剂通过毛细作用在纸上渗透扩展的过程中,待分离物质因在固定相与展开剂中的分配系数不同而得到分离。
2.2.1正相纸色谱法 正相纸色谱法是指以纸为载体,以纸上所含水分作为固定相,用展开剂展开的分配色谱法。正相纸色谱法在放化纯度测定中的应用比较广泛。ChP 2020和USP 43-NF38收录的用于甲状腺疾病诊断或治疗用碘[131I]、碘[123I]相关放射性药品,包括碘[131I]/碘[123I]化钠口服溶液、碘[131I]/碘[123I]化钠胶囊,均采用纸色谱法进行放化纯度测定,展开剂为甲醇的水溶液,放射性主峰碘[131I]/碘[123I]离子Rf约为0.8,可能存在的放射性杂质碘[131I]/碘[123I]酸根Rf约0.3,能够实现主峰与杂质峰的有效分离[5,9]。为了降低或避免碘[131I]/碘[123I]与纸纤维的特异性吸附效应,在进行供试品溶液点样前,先取一定量的载体溶液适量点于色层纸上,晾干后在同一位置进行供试品溶液的点样。USP 43-NF38采用淀粉遇碘变蓝的显色原理对碘离子进行鉴别[9]。另外,ChP 2020收录的来昔决南钐[153Sm]注射液、磷[32P]酸钠盐口服溶液、锝[99mTc]亚甲基二膦酸盐注射液等也采用了正相纸色谱法测定放化纯度。
将色层纸进行电解质溶液的预处理可以减少或消除吸附作用以提高分离效果,常用一定比例的甲醇-水或者丙酮-水、乙酸-水或乙腈-水、乙酸-乙腈-水、三氯甲烷-甲醇-水等极性大的混合溶剂体系作为展开剂。氟[18F]化钠注射液是一种正电子药物,主要用于活跃的成骨性反应骨疾病的诊断[12]。姜华等[13]开发了一种快速简便的测定其放化纯度的分析方法,即采用2%醋酸钠溶液处理过的Whatman No.1色层纸作为固定相,50%甲醇水溶液作为展开剂。氟[18F]离子与稳定的氟离子化学性质相同,配制稳定的氟离子溶液用同样的方法展开,淋洗后离子色谱分析得到的主峰位置与氟[18F]离子一致,从而实现对氟[18F]离子的鉴别。
2.2.2反相纸色谱法 以色层纸做支持体,采用低极性溶剂如烃类(石油醚、十一烷、硅油等)处理色层纸后作为固定相,再以极性大的溶剂作为展开剂时,则为反相分配纸色谱法。但目前ChP 2020、EP 10.2和USP 43-NF38均未收录该分析方法,未来在新药研发放射性药品放化纯度测定领域的应用还有待开发。
2.3 电泳法
电泳是指溶解或悬浮于电解液中带电荷的蛋白质、胶体、大分子或其他粒子,在电场作用下向其自身所带电荷相反的电极方向迁移。电泳法利用溶液中带有不同量电荷的阳离子或阴离子,在外加电场中使供试品中各组分以不同的迁移速度向对应的电极移动,实现分离并通过适宜的检测方法记录或计算。该方法准确度高,与其他方法相比,适用范围较窄、操作繁琐。一般可分为两大类:一类为自由溶液电泳或移动界面电泳,另一类为区带电泳[1]。
电泳法应用于放射性药品放化纯度测定并不广泛,只有《美国药典》和《欧洲药典》对于氯化铊[201Tl]注射液采用了区带电泳法。EP 10.2和USP 43-NF38采用醋酸纤维素薄膜作为支持介质,电解质溶液分别采用依地酸二钠溶液和戊二酸钠溶液。电泳完毕后,取出并干燥,使用合适的放射性检测器确定放射性强度分布,该品种的主要杂质是铊[201Tl](Ⅲ)离子,电泳法可有效将活性成分铊[201Tl](Ⅰ)和铊[201Tl](Ⅲ)离子分开[4-5]。
USP 43-NF38版收录的铬[51Cr]依地酸钠盐溶液也采用电泳法作为放化纯度测定方法[5]。
2.4 高效液相色谱法
高效液相色谱法是在高压下,利用不同组分与固定相和液体流动相作用力不同实现分离的色谱技术。HPLC系统包括高压输液泵、进样器、色谱柱(柱温箱)、检测器和数据处理系统[6]。
HPLC法测定放化纯度是在检测器模块处将放射性检测器串联于常规检测器(如紫外分光光度计、二极管阵列检测器、电化学检测器等)之后,色谱系统前端色谱柱与流动相相互作用实现各种组分的分离,并流经放射性检测器确定各组分的放射性分布,常规检测器在放射化学测定中的用途是检测“冷化合物”的色谱峰,并与各放射性化学成分进行比对从而实现鉴别。该方法具有准确度高、精密度好、组分之间分离效率高、适用范围广等优点,与其他方法相比,检验准备时间长、对试剂纯度要求高。
液相色谱法的分类方式很多,按HPLC法的分离原理不同进行分类,包括分配色谱法、吸附色谱法、离子交换色谱法、分子排阻色谱法等[6]。
2.4.1分配色谱法 分配色谱(partition chromatography)是最常用的液相色谱法。其分离原理是利用被分离组分在固定相和流动相中的分配性能不同实现分离。固定相为载体表面的填料,流动相为不同极性溶剂。当固定相极性大于流动相极性时,称正相色谱(normal phase high performance liquid chromatography, NP-HPLC),常用硅胶或键合极性基团的硅胶填充成的色谱柱。当固定相极性小于流动相极性时,称反相色谱(reverse phase high performance liquid chromatography, RP-HPLC)。色谱柱多以键合非极性基团的载体为填充剂填充而成。常用的填充剂有十八烷基硅烷键合硅胶、辛基硅烷键合硅胶和苯基硅烷键合硅胶等。RP-HPLC比NP-HPLC在放射性药品放化纯度测定中的应用更广泛。
177Lu-DOTATATE(标记前体分子结构示于图4)作为FDA批准的第一款多肽受体靶向放射性核素治疗(peptide receptor-radionuclide therapy, PRRT)药物[13-14],可作为患有转移性或无法手术的胃肠胰神经内分泌瘤(gastro-entero-pancreatic neuroendocrine tumors, GEP-NETs)患者的一线治疗方法[15-17]。Breeman等[18-19]报道了以C18 Symmetry色谱柱为固定相,二元溶剂体系(A=0.1%TFA水溶液,B=100%甲醇)作为流动相的RP-HPLC法。洗脱方式采用梯度洗脱,洗脱过程中能够实现在较短时间内将干扰分离的强保留杂质组分从色谱柱中清除,使色谱柱保持干净状态,以进行下一次的分析;此外,梯度洗脱具有可提高峰分离度和增加峰容量的优点[20]。
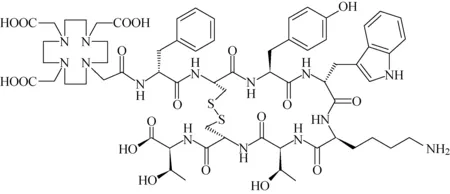
图4 DOTATATE的分子结构[18]Fig.4 The molecular structure of DOTATATE[18]
68Ga-DOTATATE可用于诊断神经内分泌肿瘤[21],Mu等[22]对比了测定68Ga-DOTATATE放化纯度的三种方法,发现采用RP-HPLC法进行梯度淋洗主峰与杂质峰分离度最好。
另外,EP 10.2收录的钴[57Co]标记的氰钴[57Co]胺胶囊/氰钴[57Co]胺溶液、钴[58Co]标记的氰钴[58Co]胺胶囊/氰钴[58Co]胺溶液、碘[131I]化钠治疗/诊断胶囊/口服溶液、[11C]雷氯必利等品种,以及USP 43-NF38收录的铟[111In]喷曲肽注射液等品种均采用RP-HPLC法作为放化纯度测定方法。
2.4.2吸附色谱法 吸附色谱法是利用被分离组分对固定相表面吸附中心吸附能力的差别进行分离。其固定相为吸附剂,流动相为不同极性溶剂,利用不同组分与固定相吸附力的不同,以及流动相对不同组分解吸附力的不同实现分离。吸附色谱属于NP-HPLC。吸附剂为多孔极性填料,常用硅胶;流动相为不同极性溶剂,常用二氯甲烷、正己烷、正丁醇、四氢呋喃等。
碘[131I]/碘[123I]苄胍注射液作为单光子显像剂(以碘[131I]苄胍注射液为例,分子结构示于图5),在诊断神经脊的肿瘤如嗜铬细胞瘤和神经母细胞等疾病方面具有较高的灵敏度和特异性。EP 10.2采用NP-HPLC分析方法,参数包括色谱用硅胶作为色谱柱的填料,80 g/L硝酸铵-稀氨水-甲醇(体积比为1∶2∶27)作为流动相,分光光度计(波长254 nm)与放射性检测器联用对主峰碘[131I]苄胍与杂质峰碘[131I]离子以及其他放射性杂质峰定性定量分析[4]。
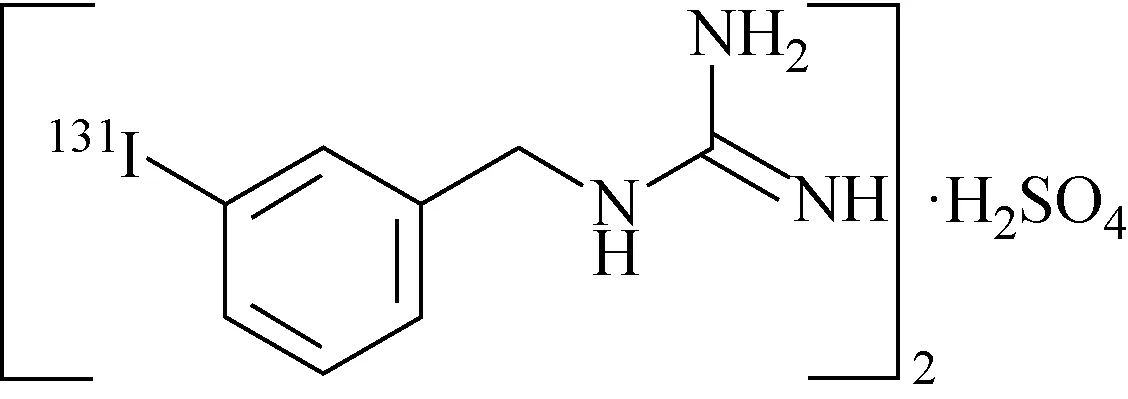
图5 碘[131I]苄胍的分子结构[4]Fig.5 The molecular structure of iobenguane[131I] sulfate[4]
2.4.3离子交换色谱法 离子交换色谱法(ion exchange chromatography, IEC)是利用被分离组分离子交换能力的差别实现分离,采用高压输液泵系统将规定的洗脱液泵入装有填充剂的色谱柱对可解离物质进行分离测定。色谱柱用离子交换填充剂填充,有阳离子交换色谱柱和阴离子交换色谱柱。
在USP 43-NF38和EP 10.2对氟[18F]化钠注射液放化纯度的测定均采用离子交换色谱法,色谱柱类型不同,其中《美国药典》中L31类型色谱柱是指季胺基改性孔径2 000 Å的交联苯乙烯和二乙烯基苯(55%)强阴离子交换树脂;另外采用的常规检测器类型也不同,USP 43-NF38采用电导检测器,EP 10.2则采用紫外检测器;流动相分别使用强酸和强碱的溶液,对色谱系统不友好,参考欧美建立标准时应慎重考虑流动相的选择。
除此以外,EP 10.2收录的氮[13N]氨水采用阳离子交换色谱法测定放化纯度,醋酸钠碳[11C]注射液采用强碱性阴离子交换法测定放化纯度。
2.4.4分子排阻色谱法 分子排阻色谱法(size exclusion chromatography, SEC)又称体积排阻色谱法,色谱柱多以亲水硅胶、凝胶或经修饰凝胶等为填充剂,因填充剂表面分布不同尺寸的孔径,样品中待分离组分进入色谱柱后,按其分子线团尺寸的差异实现分离。
《欧洲药典》收录的碘[125I]人血清白蛋白注射液放化纯度测定采用分子排阻色谱法。以尺寸排阻色谱用硅胶为色谱柱固定相,磷酸氢二钾-磷酸氢二钠-氯化钠混合溶液作为流动相,分光光度计(波长280 nm)与放射性检测器联用,并以人白蛋白溶液或其他适当的人白蛋白标准品经稀释后作为参考溶液实现对活性成分鉴别,确定放射性主峰。
2.5 多种方法联用
对于放射性化学杂质比较复杂的品种,采用单一的方法很难实现对所有杂质分离并鉴别,因此需要结合各种方法的优势制定合理的杂质分析策略。
氟[18F]脱氧葡糖注射液采用硅胶薄层色谱法对主峰2-氟[18F]-2-脱氧-D-葡萄糖与氟[18F]离子、2-[18F]氟-2-脱氧-D-葡萄糖的部分或完全乙酰化的衍生物的极性差异进行分离,而采用离子交换色谱法对杂质2-[18F]氟-2-脱氧-D-甘露糖进行测定,两种方法联用可对活性成分和杂质进行定性鉴别。
放射性核素标记的多肽类药物已越来越多应用于肿瘤、感染、血栓等的诊断和治疗,以68Ga-DOTATOC注射液为例,其活性成分是68Ga标记的多肽络合物,结构较为复杂,具体分子结构示于图6,其在临床上主要作为诊断神经内分泌肿瘤的新型药物。对其放化纯度测定采用快速硅胶薄层法分析杂质胶体镓[68Ga],采用液相色谱法对镓[68Ga](Ⅲ)离子以及其他可能存在的杂质进行分析[4]。
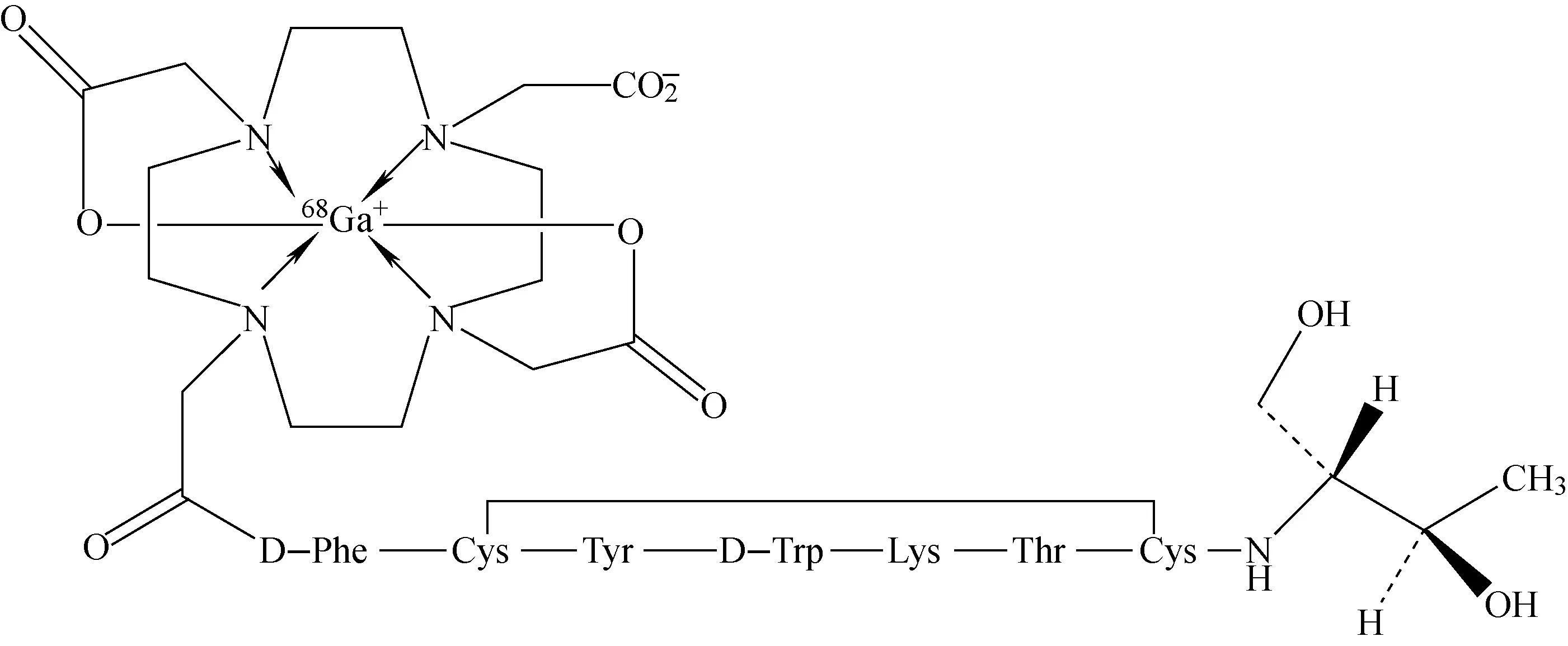
图6 68Ga-DOTATOC的分子结构[4]Fig.6 The molecular structure of 68Ga-DOTATOC[4]
多种方法联用可以针对不同的杂质采用更适宜的方法,对杂质检测的灵活性更强,适用范围更广。EP 10.2收录其他品种也采用了多种方法联用的分离技术,包括(1)18F-Alovudine注射液:正相薄层色谱法-反相液相色谱法(梯度洗脱);(2)18F-Fluromisinidazole注射液:正相薄层色谱法-反相液相色谱法;(3) L-Methionine([11C]methyl)注射液:反相薄层色谱法-反相液相色谱法;(4) 锝[99mTc]人白蛋白注射液:正相薄层色谱法-尺寸排阻液相色谱法;(5) 锝[99mTc]巯替肽注射液:正相纸色谱法-反相液相色谱法;(6) 锝[99mTc]甲氧异腈注射液:反相薄层色谱法-正相纸色谱法-反相高效液相色谱法等。
四种分析方法的优缺点比对列于表2。薄层色谱法和高效液相色谱法的适用范围最广泛,电泳法可准确地分离不同电荷的阳离子或阴离子。

表2 四种分析方法优缺点比对Table 2 Comparison of advantages and disadvantages of four analysis methods
3 总结与展望
本文较为全面地概述了国内外药典(ChP 2020、EP 10.2和USP 43-NF38)以及相关文献资料有关放化纯度测定的四种经典分析方法的特点、分离原理,并列举了应用实例。在放化纯度方法建立过程中,无论采用哪一种分析方法,需要重点关注放射化学杂质的研究。长远来看,HPLC法或与多种方法联用的分析方法将成为全面研究放射化学杂质的主流技术手段。液相色谱仪可与多种常规检测器联用,在执行的ChP 2020通则0512液相色谱法中增加了电喷雾式检测器相关内容,对拓宽HPLC法在放化纯度测定中的应用提供了更多选择。国内核医学正处于快速发展时期,放射性药品作为核医学的精神食粮,未来将会有更多新药或者仿制药开发工作的开展,如生长抑素类化合物等生物活性分子作为放射性药品的标记前体,分子结构的特殊性对放射性化学杂质研究增加了难度,尤其是在对标记后主成分与其他杂质成分的鉴别上,液相色谱仪-高分辨质谱仪联用将发挥重要的作用。
