表面改性氧化锆无机填料对齿科自酸蚀粘结剂性能的影响
2021-06-09涂宝丽张青红王海风
涂宝丽,张青红,王海风,刘 梅
(1.东华大学材料科学与工程学院,上海 201620;2.南京医科大学,江苏省口腔疾病重点实验室,南京 210029)
0 引 言
复合树脂由于具有较好的生物相容性及美观性,被广泛用于口腔修复材料领域[1]。树脂粘结剂与复合树脂配套使用,由磷酸腐蚀后涂覆向自酸蚀方向发展。自酸蚀粘结剂因为含有酸性单体不再需要对牙本质进行单独酸蚀处理,减小了发生术后敏感的概率,所以在临床中得到大量应用[2-4]。但是,其自身组分具有亲水性,通常能够与牙本质很好地结合,而与复合树脂的粘结强度不够[5]。因此,增强粘结剂与复合树脂的粘结十分必要。
无机填料的加入通常被认为可有效地增强粘结剂的性能[6]。一方面是由于无机填料比树脂基材料的的硬度高、强度高,使树脂基体可承受更多的应力,另一方面是经过活化处理的填料与牙体结合更牢固,具有应力缓冲的作用[7-8]。一般认为,填料对粘结剂力学性能的影响主要取决于填料的种类、形状和尺寸等。目前氧化锆、氧化硅和羟基磷灰石等材料已被证实能够增强粘结剂的力学性能[9]。同时,氧化锆基材料被认为是一种具有高化学稳定性的生物材料,可避免向周围组织释放有毒物质[10-11]。但是氧化锆的表面通常具有化学惰性,如果不对其表面进行预处理,不能发挥其增强力学性能的作用[12]。所以,为了对氧化锆进行硅烷偶联剂改性,可对其表面包覆氧化硅涂层[13]。Martinlinna等[14-15]报道了通过摩擦化学增加表面粗糙度,使用不同类型的硅烷偶联剂来提高氧化锆与表面氧化硅涂层的结合强度,但是该方法制备的氧化硅层不够均匀[16]。最近,溶胶-凝胶法已用于制备氧化锆表面的氧化硅涂层,以改善树脂与氧化锆之间的键合[17]。溶胶-凝胶法制备的氧化硅涂层生物相容性好,能够形成均匀的氧化硅涂层,并且涂层厚度易于调控。本研究使用溶胶-凝胶法在氧化锆表面包覆氧化硅涂层,使其能够表面改性接枝较多的偶联剂,以提高填料与树脂基体的相容性。
本文以氧化锆粉体为主要无机原料,采用溶胶-凝胶法包覆氧化硅层,并在硅烷化后按照不同比例将其添加到自酸蚀粘结剂基体中,制备了自酸蚀粘结剂。
1 实 验
1.1 原材料
单体双酚A-甲基丙烯酸缩水甘油酯(Bis-GMA)、稀释剂二甲基丙烯酸三乙二醇酯(TEGDMA)、两性单体甲基丙烯酸-2-羟乙酯(HEMA)、酸蚀剂10-(2-甲基丙烯酰氧基)磷酸单癸酯(10-MDP)、光引发剂樟脑醌(CQ)、光引发剂对二甲氨基苯甲酸乙酯(4-EDMAB)均购自Sigma-Aldrich(上海)贸易有限公司。2,6-二叔丁基-4-甲基苯酚(BHT)、正硅酸四乙酯(TEOS)、聚乙烯吡咯烷酮-K30(PVP-K30)、环己烷、正丙胺、纳米氧化锆均购自中国医药集团有限公司。γ-甲基丙烯酰氧丙基三甲氧基硅烷(γ-MPS)、亚甲基蓝购自上海泰坦科技股份有限公司。10-(2-甲基丙烯酰氧基)磷酸单癸酯(10-MDP)购自毕得医药。牙托粉购自上海奇异牙科器材有限公司,牙托水购自上海新世纪有限公司。3M/ESPE商用粘结剂、3M/ESPE复合树脂购自深圳市爱牙邦医疗器械有限公司。
1.2 材料制备
1.2.1 改性ZrO2@SiO2的制备
(1)制备ZrO2@SiO2:称取5.0 g ZrO2以及0.5 g PVP, 加入到装有0.8 mL氨水、2 mL超纯水和100 mL无水乙醇的单口烧瓶中,超声10 min使ZrO2均匀分散在液相中,然后在室温下高速搅拌30 min,迅速注入1.0 mL TEOS,继续搅拌反应8 h。离心后用乙醇洗涤,将洗涤后的沉淀物放置到120 ℃的真空烘箱中干燥24 h。
(2)ZrO2@SiO2硅烷改性:称取5.0 g ZrO2@SiO2,分别量取0.550 mL γ-MPS、0.255 mL正丙胺和100 mL环己烷,全部混合加入到250 mL单口烧瓶中,超声10 min使其分散,在室温下高速搅拌30 min,然后将所得悬浊液转移到60 ℃的油浴锅中,继续搅拌30 min完成表面改性。接着将单口烧瓶放入温度设置为80 ℃的旋转蒸发仪中除去未反应的试剂,当溶剂蒸发完毕,将其取下放入到80 ℃的真空烘箱中干燥18 h,得到改性后的ZrO2@SiO2。
1.2.2 制备树脂基体及自酸蚀粘结剂
制备树脂基体:称取30.72 g Bis-GMA和11.44 g的TEGDMA,在40 ℃下搅拌2 h使两者混合均匀,备用。
制备自酸蚀粘结剂:分别称取4.40 g、4.35 g、4.20 g、3.95 g的上述混合溶液放在20 mL的棕色小瓶中,接着分别加入0.25 g HEMA以及0.25 g 10-MDP,然后加入光引发剂10 mg CQ和40 mg 4-EDMAB,再分别加入0 g、0.05 g、0.10 g、0.25 g、0.50 g的改性ZrO2@SiO2,其加入的质量分数分别占粘结剂总量的0%、1%、2%、5%、10%。避光搅拌5 h。对照组使用3M/ESPE商用粘结剂。
其他助剂:加入质量分数占粘结剂总量10%的乙醇,溶解酸性单体10-MDP的同时也能降低粘度,有利于粘结剂对牙本质的渗透。然后加少量阻聚剂BHT,其质量占粘结剂总量的0.01%,保证粘结剂能够稳定储存。
1.3 表征及测试方法
1.3.1 场发射透射电镜(TEM)
采用场发射透射电镜(TEM,型号JEM/2100F,日本)对ZrO2、ZrO2@SiO2的晶体形貌和元素分布进行分析。
1.3.2 傅里叶红外光谱仪(FTIR)
采用傅里叶红外光谱仪(FTIR,Nicolet 8700,美国)来定性分析ZrO2@SiO2的改性效果。测试采用KBr压片法,波数范围为400~4 000 cm-1,扫描次数为32次,分辨率为4 cm-1。
1.3.3 热重分析仪(TGA)
采用热重分析仪(TGA,Q5000IR,美国)定量分析 γ-MPS在ZrO2@SiO2表面的接枝率。选择N2气氛;温度范围为30~800 ℃,升温速率为10 ℃/min。
1.3.4 机械性能测试
(1)拉伸强度测试
树脂与树脂粘结:将购买的3M/ESPE复合树脂填充到尺寸为25 mm×2 mm×2 mm的硅橡胶模具中,一次填充12.5 mm高度并光固化。将自制粘结剂均匀地涂布在树脂样条端部,用强气流吹至少5 s后光固化。最后,与上述方法相同,在涂布粘结剂的端部上将另一半树脂填充并固化。每组为8个样品,取平均值。使用万能电子材料试验机(INSTRON/5969,美国)测试粘结强度。将拉伸速度设置为0.75 mm/min,直至样品断裂,记录断裂时的最大拉伸粘结强度。
树脂和牙本质粘结:收集新鲜不含龋齿的臼齿用水彻底洗涤并干燥。将购买的牙托水和牙托粉按质量比1 ∶2 混合,待其进入粘丝期后放入尺寸为12.7 mm×9.5 mm×10 mm的预切聚乙烯管中,将牙齿垂直放置在上面。用硬组织切片机切掉牙釉质,露出牙本质并打磨表面。在牙本质上涂布粘结剂2~3层,用强气流吹5 s并光固化。将聚乙烯管放在粘合界面上,逐层填充3M/ESPE复合树脂并固化。使用硬组织切片机垂直于粘结界面分别横向、纵向切割,将其切割成尺寸为1 mm×1 mm×15 mm(粘结界面为1 mm×1 mm)的长方体样品。测试方法同上。
(2)压缩强度测试
将粘结剂填充到φ4 mm×6 mm的模具中,顶部和底部分别固化40 s,每组共制作8个样品,取平均值。使用万能电子材料试验机(INSTRON/5969,美国)测试压缩强度,设定以1 mm/min的速度加载,测得最大压缩强度。
(3)光固化深度测试
将粘结剂填充在φ4 mm×10 mm蓝色不透光模具中,使用蓝光固化灯照射上表面20 s,刮除未完全固化的部分,记录固化深度。
1.3.5 断面形貌测试
通过场发射扫描电子显微镜(FE-SEM,Hitachi S-4800,日本)拍摄断裂样品表面,断裂样品来自于力学性能测试中断裂的样条。
1.3.6 牙本质小管封闭测试
对1.3.4(1)中臼齿进行处理。在牙本质上涂布粘结剂2~3层,然后用强气流吹5 s并光固化。通过场发射扫描电子显微镜观察表面。
1.3.7 微渗漏测试
收集新鲜不含龋齿的臼齿用水彻底洗涤并干燥。将牙齿分为A、B两组,在牙齿的咬合面使用硬组织切片机钻洞来模拟龋齿,确保龋洞的深度到达牙本质。A组直接在龋洞逐层填充复合树脂并固化;B组选择具有最佳粘结强度的试验组胶粘剂,将其涂抹在龋洞中并光固化,然后逐层填充复合树脂光固化。用石蜡封闭牙根尖,在填充体周围1 mm区域外涂两层指甲油,将牙齿样本可能存在的其他缝隙封闭,防止染料进入。干燥后,将所有牙齿浸泡在质量分数为1%的亚甲基蓝溶液中24 h。浸泡完毕后,将牙齿表面残留的所有染料冲洗掉,用硬组织切片机沿颊舌方向切片,在超景深显微镜(DVM6/DVM6,中国)下以72倍的放大倍数观察切片样品,并通过场发射扫描电子显微镜拍摄切片的粘结区域。
2 结果与讨论
2.1 无机填料改性
图1是ZrO2@SiO2的TEM照片以及元素分布图,可以看出ZrO2颗粒大小为25~60 nm,SiO2层厚度约为2~3 nm,包覆层致密均匀。这是由于TEOS分子中含有可水解的乙氧基团(-OC2H5),-OC2H5能够水解成硅醇基团(Si-OH),两个硅烷醇分子缩合形成硅氧烷键(Si-O-Si),再进一步缩聚形成氧化硅网络。氧化硅表面的羟基与氧化锆有氢键作用,同时后者作为成核剂使其表面形成氧化硅层[17]。

图1 ZrO2@SiO2的透射电镜照片及元素分布图Fig.1 TEM images and element mappings of ZrO2@SiO2
图2为改性前后的ZrO2@SiO2傅里叶红外光谱,波数在3 430 cm-1左右处较宽的吸收峰是由于样品表面大量羟基的伸缩振动引起的[18]。在波数为1 640 cm-1和1 110 cm-1左右的吸收峰分别由SiO2表面含有的羟基弯曲振动所致和Si-O-Si键弯曲振动所致,γ-MPS水解产生的Si-OH与SiO2表面的Si-OH发生缩聚反应,通过化学键接枝在SiO2表面。从改性之后的ZrO2@SiO2的红外光谱中可看出,与改性前的样品相比,波数在1 718 cm-1和1 580 cm-1左右的位置出现了新的吸收峰,这分别是由硅烷偶联剂 γ-MPS中的C=O键和C=C键伸缩振动所引起的,说明改性成功[19-20]。
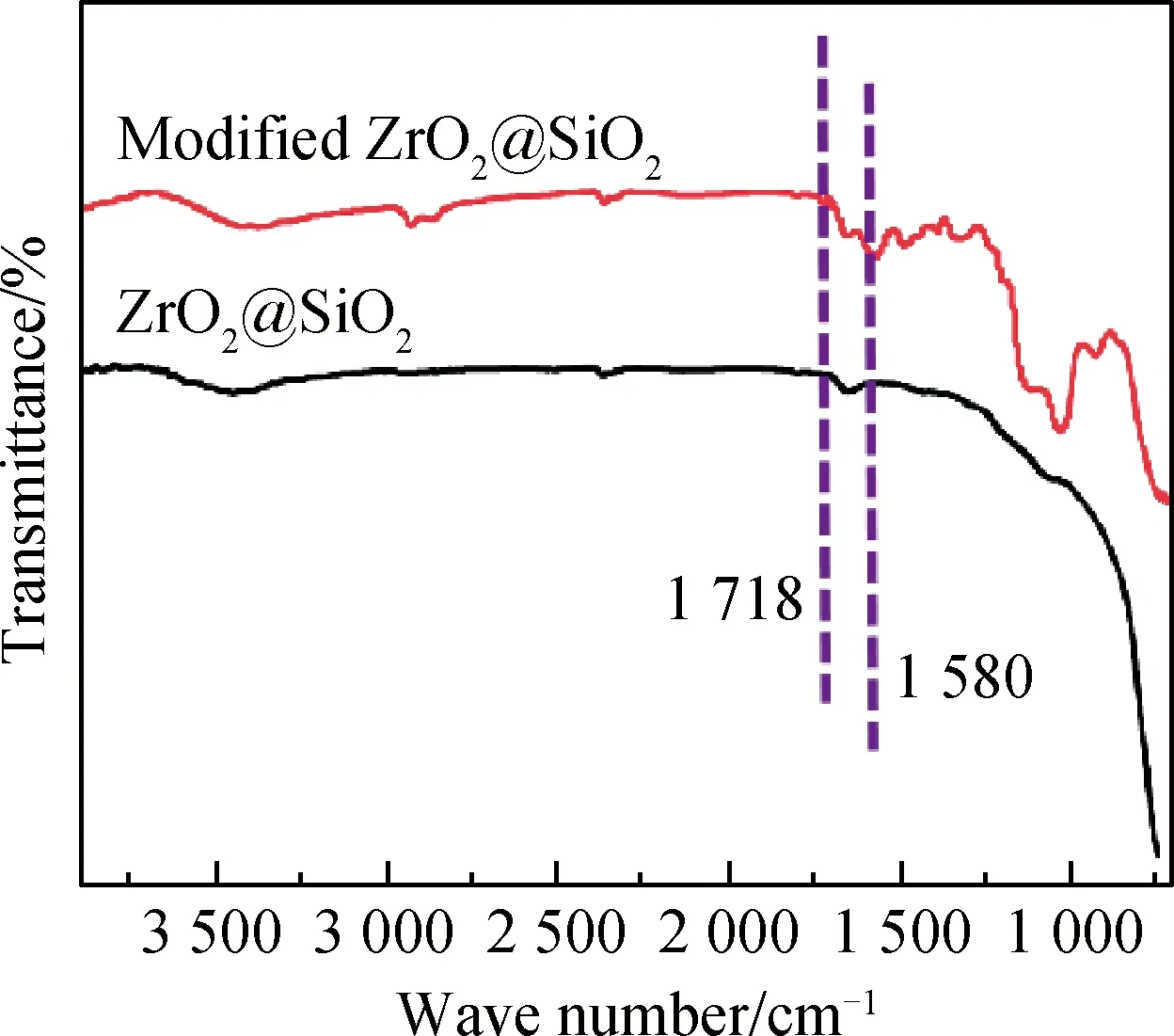
图2 改性前后ZrO2@SiO2的傅里叶红外光谱Fig.2 FTIR spectra of ZrO2@SiO2 andmodified ZrO2@SiO2
图3为ZrO2@SiO2经γ-MPS改性处理后热重曲线。热重曲线分为两个阶段:第一阶段的失重在375~558 ℃,这主要是由γ-MPS中有机基团的氧化、分解所导致的;第二阶段的失重发生在558~800 ℃,该阶段失重速度明显减慢,与SiO2中Si-OH转变为Si-O-Si有关,这部分失重的质量分数约为0.8%。因此由热重定量分析可知,ZrO2@SiO2表面的 γ-MPS接枝率约为3.1%。
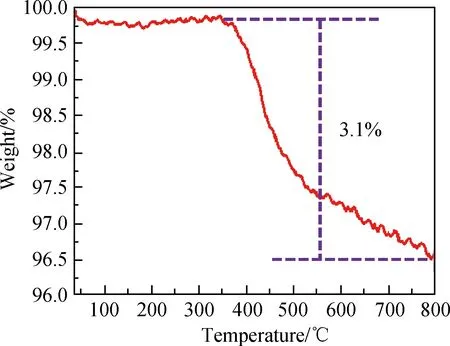
图3 改性后ZrO2@SiO2的热重曲线Fig.3 Thermogram curve of modified ZrO2@SiO2
2.2 粘结剂的力学性能测试
图4为添加了不同含量改性后的无机填料ZrO2@SiO2粘结剂的固化深度。从图中可以看出随着无机填料含量的增加,对粘结剂的透光性起不利影响,固化深度逐渐减小。当试验组填料的质量分数最高达到10%时,其固化深度仍可达到3.72 mm左右,对于粘结剂来说这个固化深度已足够。因此就固化深度单方面来考虑的话,填料质量分数为1%~10%均合适。
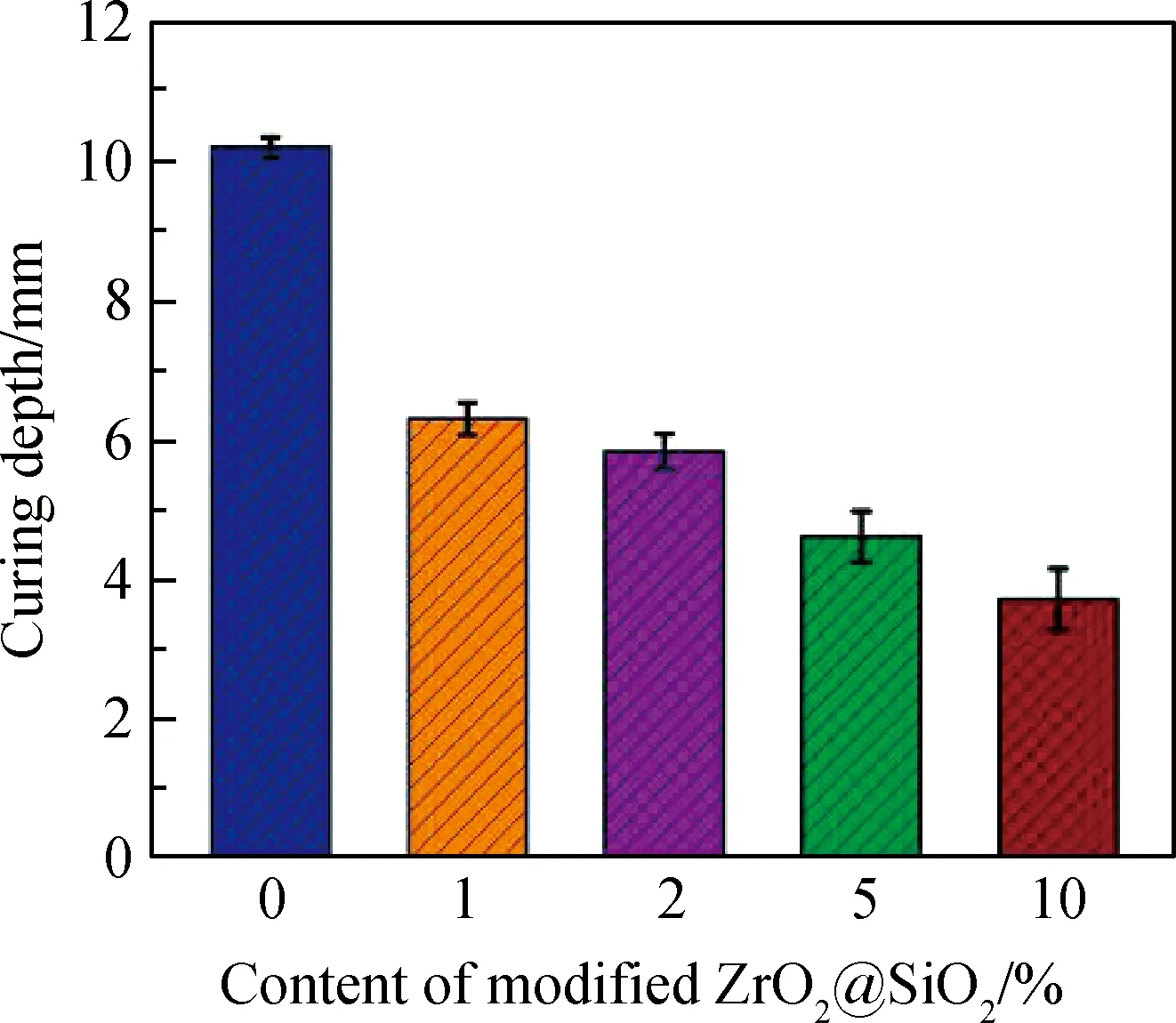
图4 不同含量改性后ZrO2@SiO2粘结剂的固化深度Fig.4 Curing depth of adhesive containing differentamounts of modified ZrO2@SiO2
图5为添加了不同含量改性后的无机填料ZrO2@SiO2粘结剂的力学性能,其中图5(a)为粘结强度,图5(b)为压缩强度。由图5可知,粘结剂的粘结强度和压缩强度都呈现出先增大后减小的趋势,当无机填料的质量分数为2%时粘结强度最高,为(31.63±1.53) MPa,当填料的质量分数为1%时压缩强度最高,为(242.10±38.30) MPa,压缩强度在填料质量分数为2%时也基本得到保持。其中,粘结强度的增强作用较明显,提高了约78.8%,压缩强度的增强作用提高了约17.5%(填料质量分数为1%)。当填料的质量分数超过5%时,粘结强度有所下降。这是由于ZrO2表面包覆的SiO2经过硅烷偶联剂改性,在光固化时与粘结剂基体形成了化学键,并且填料中ZrO2的晶态属性、Zr-O的强键能使粘结强度增高。而随着填料含量的增加,一方面是减小了粘结剂的透光性,使聚合度有所减弱从而使其力学性能减小;另一方面可能是由于无机填料发生了团聚,难以保持高度分散的状态[21]。与3M/ESPE商用粘结剂(3M)进行了力学强度的对比,结果显示商用粘结剂的粘结强度和压缩强度分别为(16.29±3.13) MPa和(67.92±16.16) MPa,所以自制的试验组粘结剂力学性能明显优于商用粘结剂。
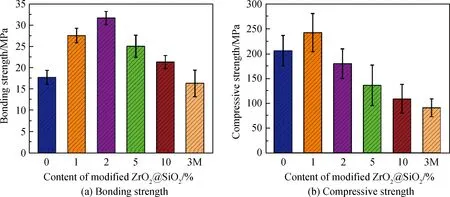
图5 不同含量改性后ZrO2@SiO2粘结剂的粘结强度及压缩强度Fig.5 Bonding strength and compressive strength of adhesive filled with different amounts of modified ZrO2@SiO2
2.3 粘结剂断面的扫描电镜照片分析
图6为粘结剂与复合树脂和牙本质的粘结断面图。图6(a)为直接切割的牙本质断面扫描电镜照片,可以看出切割后的牙本质小管被其表面存在的玷污层封闭,图6(b)为复合树脂的断面扫描电镜照片,图6(c)和图6(d)分别为含质量分数为2% ZrO2@SiO2的粘结剂对牙本质小管和复合树脂的粘结断面扫描电镜照片。由图可知自酸蚀粘结剂由于存在有机酸蚀剂可溶解牙本质表面存在的玷污层,有利于粘结剂渗入到牙本质小管中,并且其与复合树脂的粘结断面表面形成的台阶较多,起到了强度分散的作用,有利于粘结剂力学性能的提高[22]。
图6(e)和图6(f)分别为含质量分数为10% ZrO2@SiO2的粘结剂对牙本质小管和复合树脂的粘结断面扫描电镜照片,可以看出部分牙本质小管裸露,粘结剂对其的渗入作用减弱,与复合树脂的粘结表面也可看到大量的填料颗粒。推测是由于随填料含量的增加,粘结剂的粘稠度也随之增加,并且填料可能发生了团聚,无机填料过多,因此减弱了其分散强度和消耗能量的作用[23]。

图6 牙本质小管和复合树脂的断面FE-SEM照片Fig.6 FE-SEM images of fracture surfaces of dentin tubules and composite resin
2.4 牙本质表面的场发射扫描电镜照片分析
图7为牙本质表面的场发射扫描电镜(FE-SEM)照片。图7(a)为磷酸(质量分数37%H3PO4)酸蚀的牙本质表面,图7(b)为在切割后的牙本质表面涂抹粘结剂后的扫描电镜照片。从图中可以看出经过磷酸酸蚀的牙本质表面暴露出管径2~3 μm左右的牙本质小管,涂抹粘结剂的牙本质表面则未见牙本质小管暴露,与图6中的牙本质粘结断面扫描电镜对比可证明,粘结剂除了可以酸蚀掉切割牙本质表面的玷污层外,还有很好的牙本质小管封闭作用和表面浸润性。

图7 牙本质表面的FE-SEM照片Fig.7 FE-SEM images of dentin surface
2.5 微渗漏情况的显微镜照片分析
图8为使用超景深显微镜观察的微渗漏照片,图8(a)为未使用粘结剂,直接将复合树脂填充到模拟龋洞的对照组,图8(b)为选择力学性能最好的试验组粘结剂的微渗漏情况。显微镜照片显示对照组微渗漏现象非常明显,试验组仅发生了轻微渗漏。图9为充填体边缘场发射扫描电镜照片,可以看出由于粘结剂的存在明显地使充填体与牙齿之间缝隙减小,使其结合更紧密。这是由于自酸蚀粘结剂部分酸蚀掉牙本质表面的玷污层后渗入到牙本质小管中,而粘结剂中又存在两亲性成分以及硅烷偶联剂改性过的填料,有利于其与复合树脂的结合,所以复合树脂与渗入的粘结剂和部分残留的玷污层三者混合固化,可以达到粘结固位的目的[24]。

图8 超景深显微镜下的微渗漏情况Fig.8 Microleakage under ultra-depth-of-field microscopy

图9 充填体边缘场发射扫描电镜照片Fig.9 FE-SEM images of filling body edge
3 结 论
为解决氧化锆难以接枝偶联剂的难题,本文通过在其表面包覆了2~3 nm厚无定形氧化硅层成功接枝硅烷偶联剂,并将其以质量分数为1%~10%的比例添加到自酸蚀粘结剂中。适量的添加明显提高了粘结剂的力学性能,添加质量分数为2%后其粘结强度提高了约78.8%,无机填料超过5%时粘结强度有所下降。适量填料添加后,加强了粘结剂对牙本质小管的渗入作用,可明显减轻微渗漏现象,这也将在一定程度上延长了齿科修复材料的服役寿命。
