一个新颖阳离子型铱(Ⅲ)配合物的合成及其力致发光变色性能研究
2021-06-04任孟然孙日勇陈秋宏唐怀军王羽虹李浩聚李相华王凯民
任孟然,孙日勇,陈秋宏,唐怀军,王羽虹,李浩聚,李相华,王凯民
(云南民族大学化学与环境学院,云南省高校绿色化学材料重点实验室,云南 昆明650500)
力致发光变色(Mechanochromic Luminescence,MCL)材料是指在外力(剪切、拉伸、压力等)作用下,聚集状态或分子结构发生改变,发光颜色随之发生改变的化合物[1-4].这类物质在外力撤销后,通过溶剂熏蒸、加热等方式通常又可以恢复原来的发光颜色,表现出动态可逆的固态发光变化,是一类重要的智能材料(Smart Materials)[1-4],在压力传感、数据存储、光电器件、防伪和显示等领域具有重要的应用价值[1-7].
目前已经报道的力致发光变色材料,几乎都是有机类发光材料,主要包括有机小分子荧光化合物[1,3-6]、有机发光配合物[2-4,7-16]、高分子发光材料[17]等.其中以发光配合物的种类和数量最多,主要包括Au(Ⅰ)[2,4,8-9]、Ir(Ⅲ)[2,7]、Cu(Ⅰ)[2,14]、Zn(Ⅱ)[2,10-11]、B(Ⅲ)[12-13]、Pt(Ⅱ)[2,4,15-16]等元素的有机配合物.目前,进一步丰富这类发光材料的种类和数量,阐明其发光颜色变化机理,拓展其应用领域和提升相关性能等各方面的研究方兴未艾,这些研究工作具有重要的理论和实用价值.
阳离子型有机铱(Ⅲ)配合物是最近十多年迅速发展起来的一类重要的有机金属配合物发光材料,具有发光效率高(理论量子效率为100%)、亮度高、易于通过配体变换调节发光颜色、光和热稳定性高、合成产率高等优良特性[18-22],在有机发光二极管[18-19]、发光电化学池[18,20]、LEDs用下转换发光材料[18,21]、化学发光检测[22]、细胞成像[22-23]等领域被广泛应用.近年来,聚集诱导发光[24-25]、力致发光变色[2,7,25-28]等备受关注的新型发光现象在阳离子型铱(Ⅲ)配合物中也有所发现,相关研究正在不断深入.如早在2012年Shan等[26-27]就报道了一些具有力致发光变色性能的阳离子型铱(Ⅲ)配合物.同年,Mastropietro等[28]也报道了类似性能的阳离子型铱(Ⅲ)配合物.2014年Sun等[7]设计合成的数个阳离子型铱(Ⅲ)配合物,具有显著的力致发光变色效应并被成功地应用于数据存储,表现出很高的数据安全性.最近,Zhao等[25]报道了3个同时具有聚集诱导发光和力致发光变色效应的阳离子型铱(Ⅲ)配合物.不过,通过对上述文献及一些其他相关文献进行综合分析可以发现,迄今为止,具有力致发光变色性能的阳离子型铱(Ⅲ)配合物总数量上依然非常少,尤其是具有高对比性发光变色性能的此类配合物更是偏少,进一步合成、研究并开发应用此类物质颇为重要.
本工作以2-苯基吡啶(ppy)为主配体,N,N-二苯 基-4-(4-苯 基-5-(吡啶-2-基)-4H-1,2,4-三 唑-3-基)苯胺(DPPTA)为辅助配体,合成了一个新颖的具有力致发光变色性能的阳离子型铱(Ⅲ)配合物[(ppy)2Ir(DPPTA)]PF6.本文对该配合物进行了恰当的表征后,对其力致发光变色行为和可能的机理进行了研究.
1 实验部分
1.1 仪器与试剂化学药品和试剂购自上海泰坦科技股份有限公司,均为分析纯,未经进一步纯化. 德国Bruker公司AV400型核磁共振光谱仪、D8 ADVANCE A25XX射线衍射仪;美国Agilent公司1100 LC/MSD TOF液相色谱-质谱仪;北京普析UVWin 6紫外-可见吸收光谱仪;法国Jobin Yvon公司FL3-21荧光光谱仪;德国Netzsch公司DSC 200差示扫描量热仪和STA 449F3热综合分析仪.
1.2 阳离子型铱(Ⅲ)配合物的合成
1.2.1 N,N-二苯基-4-(4-苯基-5-(吡啶-2-基)-4H-1,2,4-三唑-3-基)苯胺(DPPTA)的合成 合成路线如图1所示,具体过程如下:N, N-二苯基-4,4-(5,5-(吡 啶-2,2-基)-1,3,4-噁 二 唑-2,2-基)苯 胺(DPPOA)[21]和离子液体三氟乙酸吡啶[29]均按文献合成.称取1.00 g(2.6 mmol)DPPOA、0.48 g(5.2 mmol)苯胺和2.50 g(13.0 mmol)三氟乙酸吡啶加入烧瓶中,Ar保护,110℃反应8 h.冷却后加入100 mL CH2Cl2,再水洗(50 mL×3)除去三氟乙酸吡啶,旋转蒸发除去有机溶剂,残留物过硅胶柱提纯,以石油醚-CH2Cl2(体积比1∶1)淋洗,得灰白色固体产物1.17 g(产率98.1%).1H NMR (400 MHz,CDCl)δ: 8.32(d,1H,3J=4.0 Hz,ArH), 8.07(d,1H,3J=8.0 Hz,ArH),7.73(t,1H,3J=7.6 Hz,ArH),
7.40(d,3H,3J=6.8 Hz,ArH),7.26(m,8H,ArH),7.19(t,1H,3J=6.4 Hz,ArH),7.07(t,6H,3J=8.0 Hz,ArH),
6.91(d,2H,3J=8.8 Hz,ArH).

图1 [(ppy)2Ir(DPPTA)]PF 6的合成路线Fig.1 Synthetic route of [(ppy)2Ir(DPPTA)]PF6
1.2.2 [(ppy)2Ir(DPPTA)]PF6的合成 铱(Ⅲ)二聚物(ppy)2Ir(μ-Cl)2Ir(ppy)2(ppy:2-苯基 吡 啶)按 文献[30]合成.称取1.07 g(1.0 mmol)(ppy)2Ir(μ-Cl)2Ir(ppy)2和0.93 g(2.0 mmol)DPPTA加入到40 mL乙二醇中,Ar保护,150℃搅拌反应16 h.冷却后加入10 mL1.0 mol·L-1NH4PF6溶液,过滤,水洗,干燥后得粗产物.过硅胶柱提纯,以CH2Cl2-乙腈(体积比10∶1)淋洗,旋干得黄色固体粉末1.67 g(产率75.2%).1H NMR(400 MHz,CDCl3)δ:8.15(d,1H,3J=5.6 Hz,ArH),7.91(m,3H,ArH),7.78(t,3H,3J=8.0 Hz,ArH),7.72(m,4H,ArH),7.66(m,4H,ArH),7.60(m,1H,ArH),7.32(m,1H,ArH),7.27(s,1H,ArH),7.25(d,4H,3J=7.6 Hz,ArH),7.06(m,7H,ArH),6.96(m,2H,ArH),6.90(d,1H,3J=7.4 Hz,ArH),6.8 6(t,1H,3J=7.6 Hz,ArH),6.8 0(d,2H,3J=8.8 Hz,ArH),6.3 3(t,2H,3J=7.6 Hz,ArH).MS(ESI+)m/z(C53H39F6IrN7P):计算值966.2 9[M-PF6]+,实测值966.3 [M-PF6]+.
黄色固体粉末产物再溶于少量CH2Cl2中,自然挥发至干得绿色固体产物.
2 结果与讨论
2.1 紫外-可见吸收光谱图2为[(ppy)2Ir(DPTTA)]PF6在CH2Cl2溶液(1×10-5mol·L-1)中的紫外-可见(UV-Vis)吸收光谱.[(ppy)2Ir(DPTTA)]PF6在245~385 nm之间的吸收较强,这部分吸收系由配合物的配体中自旋允许的1π-π*跃迁所引起,包含融合一起的左右两组峰.左侧峰吸收更强,最大吸收波长位于254 nm处,其摩尔吸光系数(ε)为3.8 8×104L·mol-1·cm-1,除了254 nm左右强的吸收,大致在270 nm和290 nm处还有2个弱的肩峰.右侧峰最大吸收波长为336 nm(ε=2.1 1×104L·mol-1·cm-1),吸收相对要弱.位于385~520 nm的长波长吸收在铱(Ⅲ)配合物中通常由单线态、三线态的金属离子-配体电荷迁移跃迁(1MLCT、3MLCT)和配体中自旋禁阻的3π-π*跃迁的混合吸收引起,上述3类跃迁通常不易发生,对光的吸收非常弱[18-19].但该配合物的辅助配体DPTTA中存在由供电子基团三苯胺和吸电子基团三唑组成的双极性结构单元(Bipolar unit),促进了电子的流动和迁移,导致该配合物在此波段范围内呈现出较强的吸收[19].
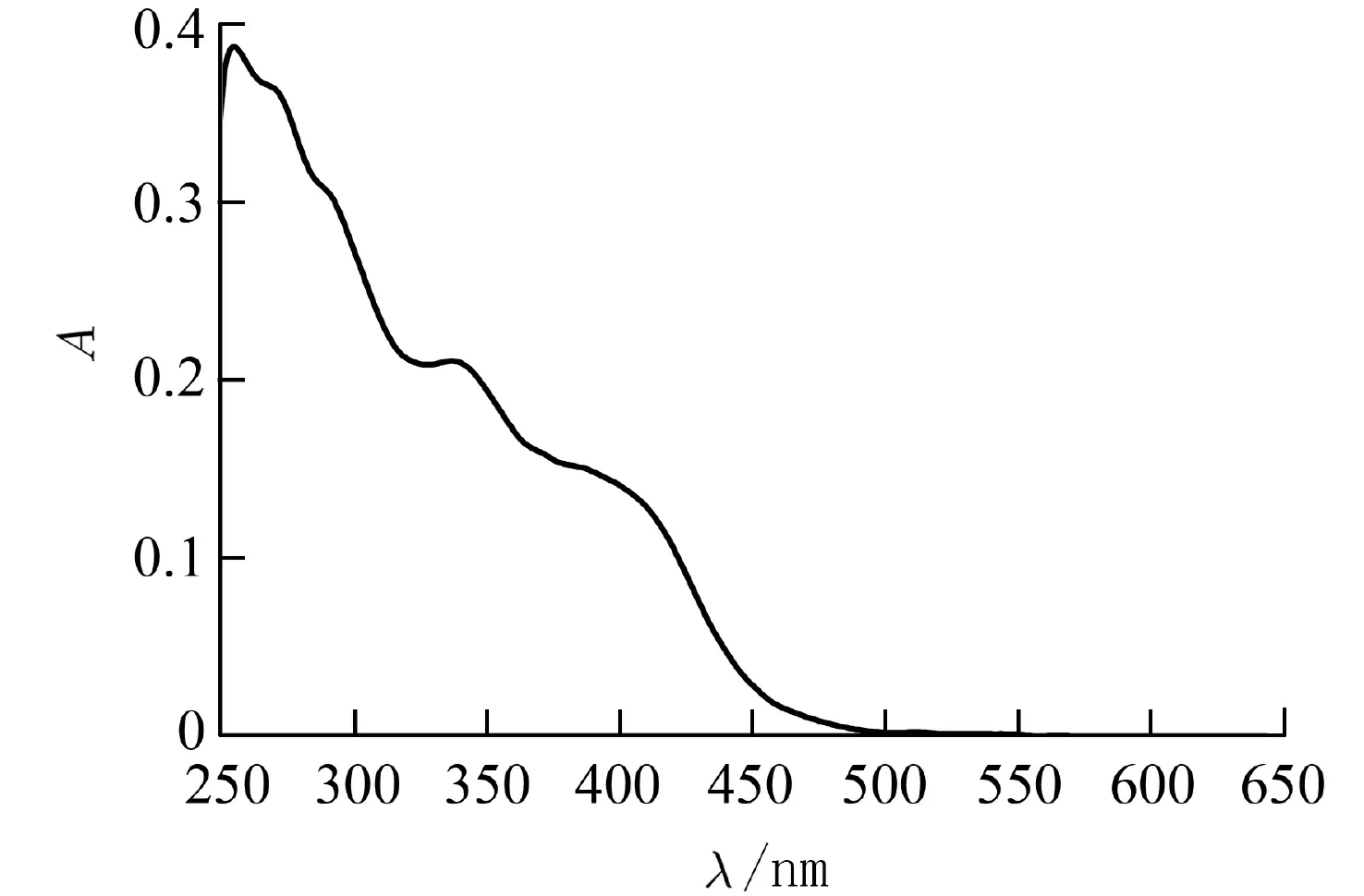
图2 [(ppy)2Ir(DPPTA)]PF6在CH 2Cl2溶液(1×10-5 mol·L-1)中的紫外-可见(UV-vis)吸收光谱Fig.2 Ultraviolet-visible(UV-vis)absorption spectrum of[(ppy)2Ir(DPPTA)]PF6 in CH2Cl2 (1×10-5 mol·L-1)
2.2 光致发光和力致发光变色性能[(ppy)2Ir(DPTTA)]PF6在CH2Cl2溶液(1×10-5mol·L-1)中的光致发光(PL)激发和发射光谱如图3所示.在CH2Cl2溶液中的激发光谱所在范围与其紫外吸收光谱基本一致,主要位于250~500 nm,但与吸收光谱相比,激发光谱右侧的强度大为提高,最大激发波长为340 nm.在CH2Cl2溶液中发射光谱位于500~700 nm之间,主要属于黄光,最大发射波长(λem,max)为567 nm.

图3 [(ppy)2Ir(DPPTA)]PF6在CH 2Cl2溶液(1×10-5 mol·L-1)中光致发光的激发光谱(λem=567 nm)和发射光谱(λex=340 nm)Fig.3 Photoluminescent(PL)excitation(λem=567 nm)and emission(λex=340 nm)spectra of [(ppy)2Ir(DPPTA)]PF6 in CH2Cl2 (1×10-5 mol·L-1)
通过挥发溶剂,从CH2Cl2溶液中析出绿色[(ppy)2Ir(DPTTA)]PF6固体,65℃真空干燥后,在蓝光(450 nm)激发下可以发出明亮的绿光(见图4),发射光谱如图5(b)中所示,λem,max为525 nm.经研磨,即受力后,转变为黄色固体粉末,在蓝光(450 nm)激发下,发射明亮的黄光(见图4),发射光谱如图5(b)所示,λem,max为576 nm,与研磨前相比,红移了51 nm. 用10 mL样品瓶盛装5 mL CH2Cl2,敞开瓶口置于100 mL烧杯底部中央,在样品瓶四周的烧杯底部撒上研磨后已变黄的[(ppy)2Ir(DPTTA)]PF6粉末,再用塑料薄膜将烧杯封住并用橡皮筋扎紧密封.可见黄色的[(ppy)2Ir(DPTTA)]PF6粉末在挥发出来的CH2Cl2蒸气熏蒸下逐渐变绿,约5 min后表面完全变为绿色.30 min后,取出样品,在蓝光(450 nm)激发下,发出明亮的黄绿光(λem,max=531 nm,见图4),与研磨后的黄光样品相比,λem,max蓝移高达45 nm,裸眼观察,颜色变化对比鲜明.其最大发射波长的变化值超过了不少文献[25-27]中同类配合物力致发光变色的最大发射波长变化值,如文献[26]中报道的2个阳离子型铱配合物,它们发光变色的λem,max差值分别为28 nm和21 nm.相比之下,可以认为[(ppy)2Ir(DPTTA)]PF6是一种高对比性的力致发光变色材料.
熏蒸后得到的黄绿光样品与最初得到的绿光样品相比(见图4和图5(b)),其发射光谱显示λem,max从525 nm红移到了531 nm,红移了6 nm,且发射峰右侧向黄光区展宽,半峰高处展宽了27 nm,说明熏蒸后样品情况与从溶液中析出所得样品情况并不完全相同(详见后面机理分析).熏蒸后黄绿光样品再经研磨后会重新发黄光,且与第1次研磨结果一致,再熏蒸再变为发黄绿光,与第1次熏蒸结果一致.研磨和熏蒸可以如此多次循环反复,各自对应状态下的发光情况稳定不变.一些文献[26-27]报道力致发光变色材料经研磨发光变色后,经加热后也可以恢复为研磨前原来的发光颜色,但[(ppy)2Ir(DPTTA)]PF6有所不同,研磨后的黄光样品经加热(从室温至200℃)后,依然保持黄色外貌和发黄光,并不恢复为绿色和发绿光或黄绿光.

图4 最初制得的[(ppy)2Ir(DPPTA)]PF6固体,经研磨后和经CH 2Cl2熏蒸后在蓝光(450 nm)激发下的发光照片及其相互转化Fig.4 PL photographs(excited by blue light,450 nm)and interconversion of as-prepared[(ppy)2Ir(DPPTA)]PF6 solid,its being ground and then fumed by CH2Cl2

图5 最初制得的[(ppy)2Ir(DPPTA)]PF 6固体,经研磨和CH 2Cl2熏蒸后的归一化的光致发光激发光谱(λem=525 nm,531 nm,575 nm)(a)和发射光谱(λex=450 nm,450 nm,450 nm)(b)Fig.5 Normalized PL(a)excitation(λem=525 nm,531 nm,575 nm)and(b)emission(λex=450 nm,450 nm,450 nm)spectra of asprepared [(ppy)2Ir(DPPTA)]PF6 solid,itsbeing ground and then fumed by CH2Cl2
3种情况下的固体样品的激发光谱(图5(a))彼此非常相似,均由2组激发峰组成,与极稀溶液下的激发光谱(见图3)相比,由于固体状态下[(ppy)2Ir(DPTTA)]PF6分子的聚集,激发光谱出现宽泛和红移.最初得到的样品和熏蒸后激发光谱几乎完全一样.但研磨后样品其激发光谱更为宽泛和红移,右侧大约从500 nm展宽到550 nm以后.
与[(ppy)2Ir(DPTTA)]PF6以极稀浓度存在于CH2Cl2溶液中发射黄光(见图3,λem,max=567 nm)相比,研磨后样品也发黄光,但更加红移(λem,max=575 nm),而从溶液中析出的固体样品(λem,max=525 nm)以及研磨后再用蒸气熏蒸后的样品(λem,max=531 nm)相对溶液中发光则是蓝移的.相比溶液中析出的固体,[(ppy)2Ir(DPTTA)]PF6在溶液中发光红移至黄光,可能与其辅助配体DPTTA中存在三苯胺-三唑双极性功能团有关.因为双极性基团会导致较强的分子内电荷迁移跃迁(Intramolecular charge transfer,ICT)[11],而 极 性 溶 剂CH2Cl2会加强这种ICT,相比几乎无溶剂的[(ppy)2Ir(DPTTA)]PF6晶体,发光更加红移.但红移程度不如研磨后的固体样品,因为研磨后分子间的堆积更加紧密,原来因空间位阻不共平面的各芳香环(如三苯胺、三唑的N原子上连接的苯环)被迫共平面(或更接近共平面),整个分子中π电子共轭体系变大,导致其激发光谱和发射光谱红移.另外,分子间的π-π堆积程度也会因力的作用而提高,这也会导致激发光谱和发射光谱的红移.上述现象在其他一些力致发光变色材料的研究中,也有相似的报道和分析[11,26].
2.3 力致发光变色机理力致发光变色现象主要是由聚集状态或分子结构发生改变所致,前者更为常见.为探索[(ppy)2Ir(DPTTA)]PF6的力致发光变色机理,对上述3种情况下获得的样品进行了X射线衍射(XRD)分析和热分析.图6为3种样品的XRD图谱,从图6可知,[(ppy)2Ir(DPTTA)]PF6最初为分子排列有序的结晶状态,经研磨后,在力的作用下,转变为混乱无序的无定形态.用CH2Cl2蒸气熏蒸后,[(ppy)2Ir(DPTTA)]PF6重新结晶,但与从溶液中析出的晶体相比,CH2Cl2熏蒸后再结晶产物的XRD图谱虽然与前者的XRD图谱较为接近,但部分衍射峰的位置和强度存在一定的差异,应该是其结晶程度、晶粒大小及形态、分子聚集状态等应与从溶液中析出有所不同所致,在其他报道[24-26]中也存在类似的情况.

图6 最初制得的、经研磨和CH 2Cl2熏蒸后的[(ppy)2 Ir(DPPTA)]PF6固体的XRD图谱Fig.6 XRD patterns of as-prepared[(ppy)2Ir(DPPTA)]PF6 solid,its being ground and then fumed by CH2Cl2
从上述样品的热分析(DSC和TG,10℃·min-1,N2气氛,图7)结果来看,从室温至200℃(主要发生在150℃以前)时均经历了吸附的溶剂和水等的失重过程.2种结晶的样品,即最初制得的和熏蒸后得到的样品到200 ℃时均失重约4.9%,但失重过程有所差异,最初溶液中结晶析出得到的样品可能结晶得更加完好和致密,失重更延后一点,室温加热至约70℃以后才显著脱附,而熏蒸后结晶样品从一开始升温即明显脱附.经研磨的无定形产物也是从一开始升温即明显脱附,但至200℃时失重约3.3%,吸附的溶剂量明显不如2种结晶状态.200~300℃之间,TG显示上述3种样品均无明显的失重,但DSC显示3种样品均经历一次放热过程,最初制得的、研磨后和熏蒸后样品的放热峰值分别为268℃,256℃和254℃,而2种结晶样品在放热之前还经历了一次吸热过程,最初制得的和熏蒸后的样品吸热峰值分别为244℃和222℃.上述情况说明3种样品在200~300℃之间均无明显的脱附、分解等失重发生,但存在相变.对2种晶体样品而言,先分别在244℃和222℃左右发生了晶体向无定形的转化(吸热),然后再次分别在268℃和254 ℃左右发生结晶(放热),而研磨得到的无定形样品仅在256℃左右出现结晶放热过程.虽是同一种物质,但2种结晶样品的吸热峰,3种样品的放热峰,它们位置接近却又不一致,是样品存在状态(如聚集状态、颗粒大小、形貌等)不同所致.DSC和TG联合分析可以判断,到310℃以后,3种样品均逐渐出现了热分解,最初制得的、研磨后的和熏蒸后的样品的热分解温度(td)分别为316℃,334℃和337℃.

图7 最初制得的、经研磨和CH 2Cl2熏蒸后的[(ppy)2Ir(DPPTA)]PF 6固体的DSC和TG图Fig.7 DSCandTGcurvesof as-prepared [(ppy)2Ir(DPPTA)]PF6solid,its being ground and then fumed by CH2Cl2
从图7(a)可知,研磨后样品经加热再次结晶的温度高达256℃,且此时依赖升温结晶的晶体与最初溶液析出制得的以及熏蒸得到的晶体并不相同.所以,一些文献[26-27]中报道的力致发光变色材料通过较低温度(< 200℃)的加热即可还原发光颜色的方式并不适用于[(ppy)2Ir(DPTTA)]PF6(如图4所示).[(ppy)2Ir(DPTTA)]PF6这种对温度并不敏感的力致发光变色材料在某些发热情况下(如摩擦发热时的摩擦力传感)的应用可能更具有优势.
3 结论
本文合成了一个具有力致发光变色性能的新颖阳离子型铱(Ⅲ)配合物发光材料,该配合物力致发光变色属于聚集状态改变所致,其发光变色对比性高,研磨后和CH2Cl2熏蒸后最大发射波长相差高达45 nm.其力致发光变色行为对受力和溶剂熏蒸敏感,但对温度并不敏感,是具有一定应用前景的力致发光变色材料.
