Construction of a high-density adzuki bean genetic map and evaluation of its utility based on a QTL analysis of seed size
2021-06-02WANGLixiaWANGJieLUOGaolingYUANXingxingGONGDanHULiangliangWANGSuhuaCHENHonglinCHENXinCHENGXuzhen
WANG Li-xia,WANG Jie,LUO Gao-ling,YUAN Xing-xing,GONG Dan,HU Liang-liang,WANG Suhua,CHEN Hong-lin,CHEN Xin,CHENG Xu-zhen
1 Institute of Crop Sciences,Chinese Academy of Agricultural Sciences,Beijing 100081,P.R.China
2 Institute of Rice Research,Guangxi Academy of Agricultural Sciences,Nanning 530007,P.R.China
3 Institute of Economic Crops,Jiangsu Academy of Agricultural Sciences,Nanjing 210014,P.R.China
Abstract Adzuki bean (Vigna angularis (Willd.) Ohwi &Ohashi) is an annual cultivated leguminous crop commonly grown in Asia and consumed worldwide.However,there has been limited research regarding adzuki bean genetics,which has prevented the efficient application of genes during breeding.In the present study,we constructed a high-density genetic map based on whole genome re-sequencing technology and validated its utility by mining QTLs related to seed size.Moreover,we analyzed the sequences flanking insertions/deletions (InDels) to develop a set of PCR-based markers useful for characterizing adzuki bean genetics.A total of 2 904 markers were mapped to 11 linkage groups (LGs).The total length of the map was 1 365.0 cM,with an average distance between markers of 0.47 cM.Among the LGs,the number of markers ranged from 208 (LG7) to 397 (LG1) and the total distance ranged from 97.4 cM (LG9) to 155.6 cM (LG1).Twelve QTLs related to seed size were identified using the constructed map.The two major QTLs in LG2 and LG9 explained 22.1 and 18.8% of the total phenotypic variation,respectively.Ten minor QTLs in LG4,LG5 and LG6 explained 3.0–10.4% of the total phenotypic variation.A total of 9 718 primer pairs were designed based on the sequences flanking InDels.Among the 200 selected primer pairs,75 revealed polymorphisms in 24 adzuki bean germplasms.The genetic map constructed in this study will be useful for screening genes related to other traits.Furthermore,the QTL analysis of seed size and the novel markers described herein may be relevant for future molecular investigations of adzuki bean and will be useful for exploiting the mechanisms underlying legume seed development.
Keywords:Vigna angularis,genetic map,QTL,seed size,PCR-based marker development
1.Introduction
Adzuki bean (Vigna angularis(Willd.) Ohwi &Ohashi),which is sometimes called azuki bean,is an annual cultivated leguminous crop belonging to the subgenusCeratotropis.It is a self-pollinating diploid plant species with 22 chromosomes (2n=2x=22),with an estimated genome size of about 500 Mb (Kanget al.2015;Yanget al.2015;Sakaiet al.2016).Because adzuki bean has traditionally been cultivated in China,Japan and Korea,it has often been referred to as an Asian legume,but it is now grown in more than 30 countries (Tomookaet al.2003;Liet al.2017).Adzuki bean can be consumed in many forms,such as in soups,bean paste and bean stuffing.The identification of the functional components of adzuki bean (Yaoet al.2011) will likely increase the demand for this crop.Thus,new high-yielding cultivars that produce high-quality beans must be developed.
Mapping and gene mining form the basis of genetic and genomic studies;however,owing to the limited number of available molecular markers,previous researchers have only been able to construct low-density genetic maps for adzuki bean (Kagaet al.1996;Hanet al.2005;Isemuraet al.2007;Luoet al.2013).The release of the adzuki bean whole genome sequence (Kanget al.2015;Yanget al.2015;Sakaiet al.2016) enabled the construction of high-density single nucleotide polymorphism (SNP)-based genetic maps (Liuet al.2016) useful for precisely locating QTLs for specific traits (Liet al.2017).However,detecting and applying SNP markers involves a complex and fairly expensive process,and sharing SNP maps among laboratories can be difficult.Therefore,we constructed an SNP map using an F2population derived from a cross between a phenotypically diverse cultivated adzuki bean and a wild genotype.The quality of the constructed map was evaluated based on an analysis of QTLs for seed size.Additionally,a set of PCR-based markers was also developed according to the sequences flanking insertions/deletions (InDels).The SNP map,the QTL analysis and the novel markers described herein may be useful for further characterizing adzuki bean genetics.
2.Materials and methods
2.1.Plant materials and DNA preparation
An F2population comprising 143 individuals derived from a cross between a cultivated adzuki bean (Vigna angularis,P1) and a wild genotype (V.angularisvar.nipponensis,P2) was used for genetic mapping.The F2population and both parents were planted in the summer of 2018 at Guangxi,China (22.82°N,108.33°E).Young leaves were collected at the seedling stage,after which DNA was extracted with a plant DNA isolation kit (Tiangen,Beijing,China).The DNA quality and purity were checked with the ND-1000 spectrophotometer (NanoDrop,Wilmington,DE,USA),whereas the concentration was determined with the Qubit®DNA Assay Kit and the Qubit®2.0 Fluorometer (Life Technologies,CA,USA).For each sample,1.5 μg purified DNA was stored at −20°C until analysis.
2.2.Phenotype evaluation
The cultivated parent produced red and large seeds,in contrast to the brown and small seeds of the wild genotype parent.The simplest and most direct method for evaluating seed size involves measuring the 100-seed weight.Consequently,100 healthy and fully mature seeds were collected from each harvested F2plant and weighed.
2.3.Library preparation and Illumina sequencing
Sequencing libraries were constructed with the TruSeq Library Construction Kit (Illumina,CA,USA).Briefly,for each sample,1.5 μg genomic DNA was sonicated (Covaris,Inc.,USA) to generate 350-bp fragments.A poly-A tail was added to the fragments,which were then ligated to a full-length adapter.After PCR amplification and purification of the products,the size distribution was assessed with the Agilent 2100 Bioanalyzer (Agilent Technologies,USA).The target fragments were quantified based on real-time PCR.The prepared libraries were sequenced with the Illumina PE150 System and 150-bp paired-end reads were generated with an insert size of approximately 350 bp.
2.4.Sequencing data quality control and analysis
Raw reads in the fasta format were first processed with a series of quality control in-house C scripts to ensure the reliability of the reads and to eliminate base-calling duplication,adapter contamination and other artificial biases.Specifically,reads with ≥10% unidentified nucleotides,>50%low-quality bases or >10 nucleotides that aligned with the adapter were removed.Additionally,≤10% mismatches were allowed,but putative duplicates generated by the PCR during library construction were discarded.
2.5.Mapping to the reference genome and detecting SNPs
The sequenced adzuki bean genome which was recently released served as the reference genome (Sakaiet al.2016).The clean reads for each sample were aligned to the reference genome with the Burrows-Wheeler Aligner (Li and Durbin 2009).The alignment files were converted to BAM files using SAMtools Software (Liet al.2009).Additionally,potential PCR duplications were removed with the SAMtools“rmdup”command.If multiple read pairs had identical external coordinates,only the pair with the highest mapping quality was retained.The GATK Program was used to call variants for all samples (McKennaet al.2010),whereas a Perl script was applied to detect SNPs (Sunet al.2013).Moreover,ANNOVAR was used to annotate SNPs based on the GFF3 files for the reference genome.
2.6.Genotyping and detecting effective markers
Only SNP markers that were homozygous in both parents and polymorphic between parents were used for further analyses.However,markers with low coverage and obviously distorted segregation were eliminated before the linkage analysis.For example,SNPs with an abnormal base and distorted segregation (P<0.001) and integrity >60%were eliminated.The final markers between parents were classified into eight segregation patterns (ab×cd,ef×eg,hk×hk,lm×ll,nn×np,aa×bb,ab×cc,and cc×ab).Only markers with the aa×bb pattern were used to construct genetic maps.After filling and correcting all effective SNPs,those from the reference genome were classified into bins based on the recombination in the F2population.The SNPs within 10-kb intervals in a parent were considered as a bin.
2.7.Genetic map construction and QTL analysis
The bin markers were partitioned primarily into linkage groups (LGs) based on their locations on the adzuki bean reference genome (Sakaiet al.2016).The SNP markers from scaffolds were incorporated into each LG.A linkage map was constructed with the Lep-MAP Program (LOD set between 2 and 40).Genetic distances were estimated with Kosambi’s mapping function.Additionally,JoinMap4.0 was used to sort the markers into LGs,after which the LGs were assigned to their corresponding chromosomes according to the physical positions of their markers.
The inclusive composite interval mapping method was used to analyze QTLs (Liet al.2007).A significant LOD threshold for the QTLs of each trait was determined with a 3 000 permutations test (P=0.01).If there were no mapping regions associated with this LOD value,the LOD was set to 3.
2.8.Primer design and validation
On the basis of InDel positions,primers were designed with the Primer3 package (Rozen and Skaletsky 2000) and validated with adzuki bean genomic DNA.To enhance the analysis of DNA in stained gels,only InDels >4 bp were considered.The PCR amplification was completed in a 20-μL reaction volume containing 10 mmol L–1Tris-HCl,50 mmol L–1KCl,2 mmol L–1MgCl2,100 mmol L–1each dNTP,0.4 mmol L–1forward and reverse primers,20 ng genomic DNA,and 1 UTaqDNA polymerase.The PCR program was as follows:35 cycles of 94°C for 30 s,suitable temperature for primer annealing for 30 s,and 72°C for 30 s.The annealing temperature varied depending on the primer pair sequences.The PCR products were analyzed by 6% polyacrylamide gel electrophoresis and silver staining(Ayanaet al.2000).Fragment sizes were estimated based on a DNA ladder.
3.Results
3.1.Analysis of seed size
The 100-seed weights for the cultivated and wild parents were 16.5 and 3.4 g,respectively.The 100-seed weight for the F2population was 3.15–12.15 g,with an average of 5.56 g,and the frequency distribution was consistent with that of a normal quantitative trait (Fig.1-B),which suggests that seed size is a classical quantitative trait and might be controlled by multiple genes.
3.2.Sequencing data and alignment to the reference genome
After a quality control step,6.61 (P1) and 6.10 (P2) Gb clean data were generated for the two parents.The clean data for the F2population ranged from 3.0 to 5.4 Gb,with an average of 4.1 Gb.The Q30ratios (bases with a quality score of 30,indicating a 1% chance of an error and thus 99% confidence)for the parents were 93.78% (P1) and 92.72% (P2),whereas for the F2population,they ranged from 89.4 to 93.9%,with an average of 92.6%.The guanine-cytosine (GC) contents for the parents were 36.38% (P1) and 37.61% (P2).Similarly,the GC contents of the F2population ranged from 35.56 to 37.72%,with an average of 36.21% (Fig.2).
The alignment of the clean reads to the reference genome revealed the final mapping rates for the parents were 97.7%(P1) and 97.0% (P2),with an average depth of about 10×.The mapping rates for the F2individuals ranged from 91.9 to 97.7%,with a depth between 3.7× and 7.3× (average of 5.5×).
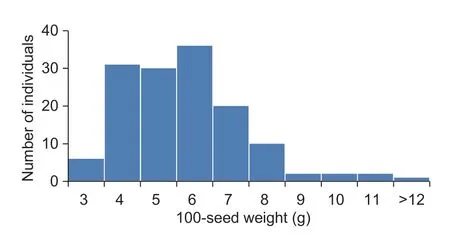
Fig.1 Frequency distribution of the seed coat color and 100-seed weight within the F2 population derived from cultivated and wild adzuki bean parents.
A total of 460 152 (P1) and 592 610 (P2) SNPs were detected in the chromosomes and scaffolds of the parents,and 545 733 homozygous SNPs were polymorphic between the parents.Additionally,262 526 SNPs had a segregation pattern of aa×bb in the F2population.After eliminating the SNPs with a sequencing depth <10× and obvious distorted segregations,the polymorphic SNPs identified on chromosomes 1–11 were classified into 2 762 bins,with an additional 284 SNP markers assigned to 154 scaffolds.These bins and SNPs were used for the linkage analysis.
3.3.Genetic linkage map construction
A total of 2 620 bin clusters from the reference chromosomes and 284 SNPs from the scaffolds were mapped to 11 linkage groups (Fig.3).Thus,the final map comprised 2 904 markers (Table 1).The number of markers per LG varied from 208 to 397,with an average of 264.The rank order of the LGs was as follows:LG1 (397)>LG9(300)>LG8 (279)>LG11 (266)>LG3 (260)>LG2 (245)>LG5 and LG4 (244)>LG10 (239)>LG6 (222)>LG7 (208).The total map length was 1 365.0 cM,ranging from 97.4 to 155.6 cM per LG,with an average length of 124.1 cM.The rank order of the LG genetic lengths was as follows:LG1 (155.6 cM)>LG3 (141.6 cM)>LG4 (138.7 cM)>LG6 (137.9 cM)>LG8 (129.5 cM)>LG7 (124.2 cM)>LG2 (116.1 cM)>LG5 (115.4 cM)>LG10 (107.4 cM)>LG11 (101.1 cM)>LG9 (97.4 cM).Additionally,LG1 and LG11 had the highest marker densities,with an average distance between markers <0.40 cM,whereas LG6 and LG7 had the lowest marker densities,with an average distance between markers >0.60 cM (Table 1).
The map included seven gaps (>5.0 cM),located in LG6 (one gap),LG7 (three gaps),LG9 (two gaps),and LG11 (one gap).The three gaps in LG7 (Block2589–Scaffold_0054,Scaffold_0108–Scaffold_1634 and Scaffold_0113–Scaffold_0208) were almost linked and spanned 30.8 cM with eight markers.The two gaps in LG9 between Block3262 and Scaffold_0255 were also almost linked,spanning 12.9 cM.
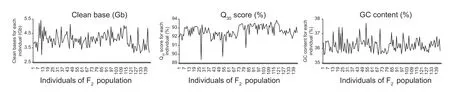
Fig.2 Distributions of clean bases (A),Q30 score (%) (bases with a quality score of 30,indicating a 1% chance of an error and thus 99% confidence) (B) and guanine-cytosine (GC) content (%) (C) for an F2 adzuki bean population.
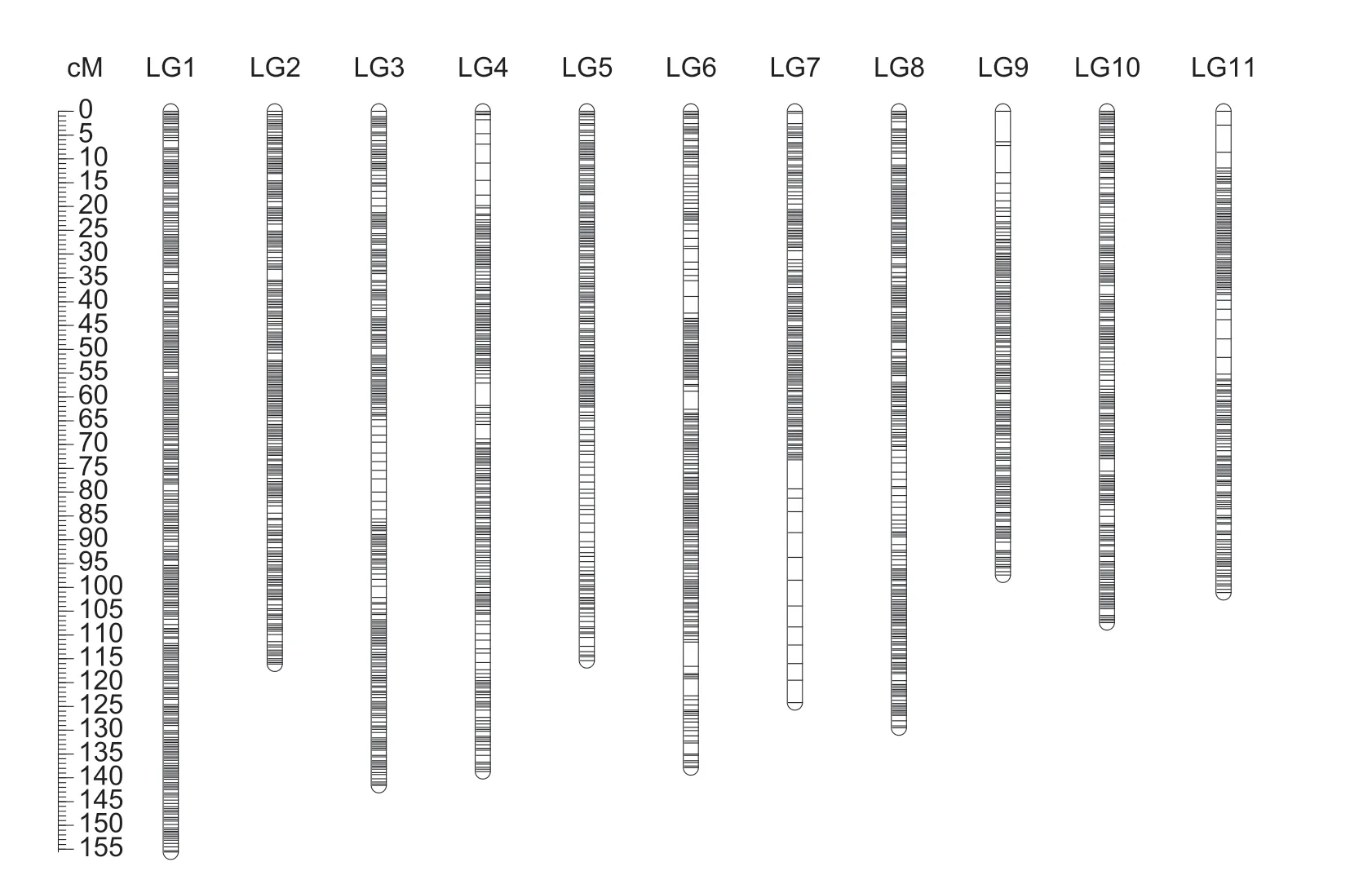
Fig.3 High-density genetic map of Vigna angularis.Map distances based on the Kosambi map unit (cM) are provided on the left.
3.4.QTL analysis and gene annotation
A total of 12 QTLs for seed size were identified,with two major QTLs on LG2 and LG9 (Fig.4).The larger one,spanning 1.46 cM and explaining 22.1% of the phenotypic variation,was on LG2 between SNP markers M129506 and M130661,with LOD=6.4.The other major QTL,spanning 0.61 cM and explaining 18.8% of the phenotypic variation,was on LG9 between markers M650027 and M650418,with LOD=6.0.The 10 minor QTLs scattered among LG4,LG5 and LG6 explained 3.0–10.4% of the phenotypic variation.All of the QTLs from the cultivated parent had an additive effect on seed size.Moreover,all of the QTLs from the cultivated parent exhibited dominance regarding seed size,with the exception of one QTL on LG9.
Twenty-two and 13 genes within the physical regions of the two major QTLs on LG2 and LG9 were physically located at 36 927 844–37 237 949 bp on chromosome 2 and 16 775 100–17 192 631 bp on chromosome 9,respectively(Appendix A).Of these genes,two were non-protein coding genes,four were predicted to encode hypothetical proteins,16 were similar to genes encoding uncharacterized proteins,and 13 were similar to functional genes from different species.
3.5.Development and validation of InDel markers
A total of 9 718 primer pairs were designed based on the sequences flanking InDels,of which 200 primer pairs expected to amplify fragments 200–215 bp long were selected and synthesized for PCR analysis.The resulting amplicons revealed that 185 of the primer pairs generated a single stable fragment of approximately 200–215 bp long,as expected,and 75 revealed polymorphisms among 24 adzuki bean cultivars (Table 2).Each locus comprised two alleles with PIC values ranging from 0.08 to 0.68,with an average of 0.38.
4.Discussion
There are different methods for constructing SNP-based genetic maps,including restriction site-associated DNA sequencing (RAD-seq) (Xuet al.2018),genotyping-bysequencing (Elshireet al.2011),specific length amplifiedfragment sequencing (SLAF-seq) (Sunet al.2013),and even whole genome re-sequencing (Liet al.2015).In adzuki bean,two SNP maps have been reported,with one constructed with RAD-seq data (Liet al.2017) and the other developed with SLAF-seq data (Liuet al.2016).These two maps,along with earlier maps constructed with traditional markers,revealed whole map lengths ranging from around 1 000 to 1 500 cM (Kagaet al.1996;Hanet al.2005;Liuet al.2016;Liet al.2017),with considerable differences in the numbers of markers,implying a similar rate of chromosomal recombination in adzuki bean.The average distance between bin markers in our map is 0.47 cM,and if converted to SNP markers,the average distance between markers decreases to only 0.13 cM.The LGs were not easily assigned to chromosomes in other maps,despite the fact the same reference genome was used (Liuet al.2016;Liet al.2017).Conversely,the LGs identified in the current study were efficiently localized to chromosomes,suggesting we constructed a high-quality map useful for future genetic and genomic investigations of adzuki bean.Moreover,we mapped a set of scaffolds to the LGs,which may help fill the gaps in the reference genome for future investigations.
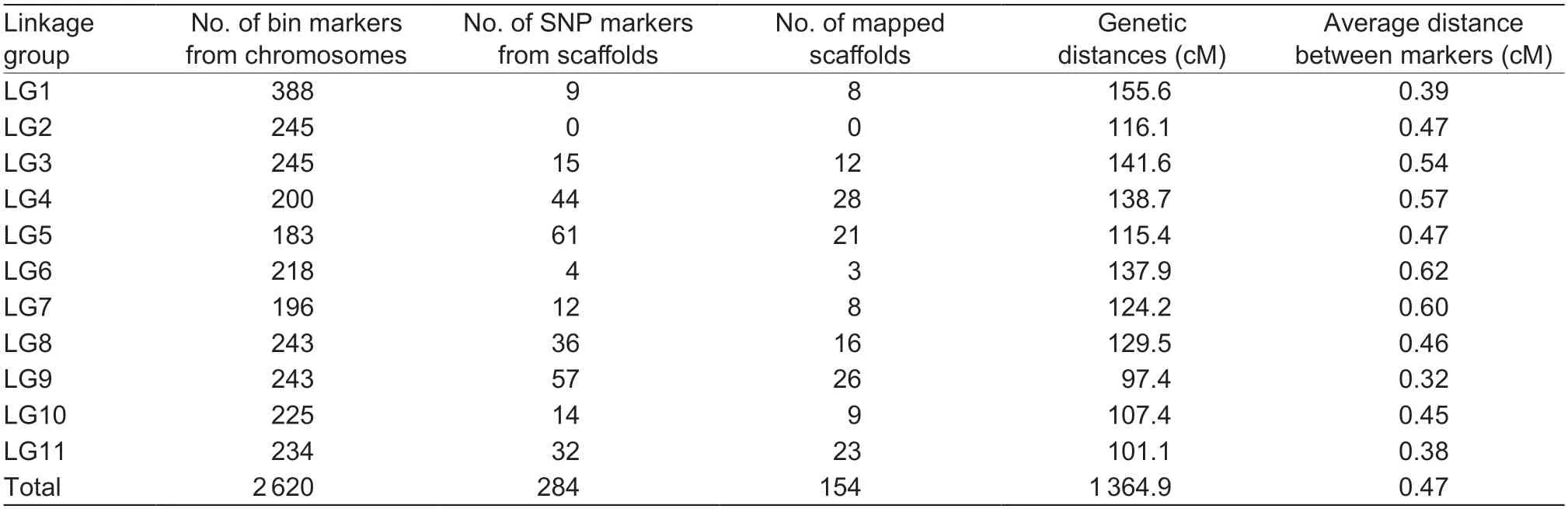
Table 1 Number of markers and integrated scaffolds,as well as the genetic distances,in an adzuki bean linkage map
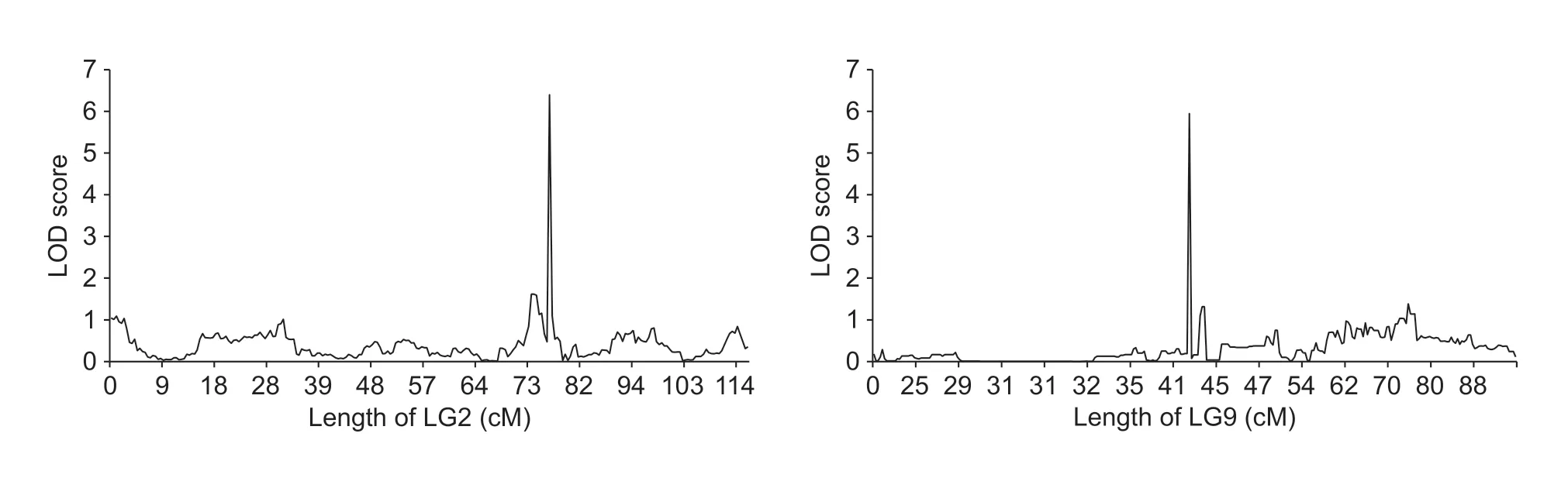
Fig.4 Major QTLs related to seed weight identified in LG2 and LG9 in adzuki bean.LOD,likelihood of odds.
(Continued on next page)
We validated the utility and quality of the map by mining for QTLs for seed size.There are some reports describing investigations on the inheritance of adzuki bean seed size(Kagaet al.1996;Jinet al.2006) as well as QTL analyses(Isemuraet al.2007;Kagaet al.2008;Liet al.2017).In the current study,we used the newly constructed genetic map to identify 12 QTLs related to seed size,which were in LG2,LG4,LG5,LG6,and LG9.This distribution pattern is similar to those in earlier studies by Isemuraet al.(2007) and Kagaet al.(2008),in which QTLs were also distributed throughout the genome.Conversely,Liet al.(2017) detected only two QTLs for seed size based on their SNP map.This difference among studies may be due to the differences in the examined materials and reference genomes and,most importantly,the effects of environmental conditions on seed size.The consistency in the QTLs identified based on our data and those detected in previous studies reflects the high quality of our map.Our genetic map may be useful for future investigations of other adzuki bean traits because there are substantial trait differences between the two parents we used.We detected two major QTLs related to seed size,with 13 genes similar to functional genes in other species within the target regions.Although seed size is an important yield component,the molecular mechanisms underlying adzuki bean seed development remain unclear.Additionally,seed size is influenced by the interactions between the genotype and environmental conditions (Renet al.2019).Identifying the most appropriate candidate genes for seed size and clarifying the mechanisms mediating their effects on seed development will require considerably more work because of the limited availability of adzuki bean genetic information.However,the results of this study will enhance future research on specific traits and may be applicable for breeding based on marker-assisted selection.
Simple sequence repeat (SSR) markers have been applied for the mapping of the adzuki bean genome (Isemuraet al.2007;Kagaet al.2008;Isemuraet al.2011) and for analyzing genetic diversity (Wanget al.2009,2012).Owing to the limited number of polymorphic SSR markers,SNP markers have gradually become more popular,especially with the development of high-throughput sequencing technology (Liuet al.2016;Liet al.2017).Specifically,the considerable density of SNP markers may enable fine mapping.However,the detection and application of SNP markers remain expensive,making them a less feasible option than PCR-based markers.Moreover,the biggest disadvantage of SNP markers is that they cannot be easily shared among laboratories.Accordingly,PCRbased markers may be better suited for most researchers and most routine experiments.The InDels detected from high-throughput sequencing usually differ by more than 5 bp between accessions,making them applicable for constructing genetic maps (Millset al.2006;Liet al.2015)and developing PCR-based markers (Moghaddamet al.2014;Daset al.2015;Zhouet al.2015).To the best of our knowledge,there are no reports regarding InDel marker development for adzuki bean.In the current study,we designed thousands of primer pairs based on the sequences flanking InDels.We selected 200 primer pairs according to the expected amplicon size for a PCR analysis.Most of these primers stably amplified the expected fragments,implying the InDel mining was highly accurate.However,only 75 primer pairs revealed variations among 24 germplasms,which may reflect the limited genomic diversity indicated by SSR markers (Wanget al.2009,2012).In future studies,we will continue to validate additional primer pairs in order to generate more markers applicable for adzuki bean genetic research.The map and associated data presented herein may be relevant for future investigations that focus on the diversity and genetic characterization of adzuki bean and related species.
5.Conclusion
A high-density genetic map of adzuki bean was constructed,with an average distance between bin markers of only 0.47 cM.Two major and 10 minor QTLs for seed size were identified,indicative of the high quality of the constructed map.Additionally,a set of PCR-based markers was developed according to the sequences flanking InDels.This map and the results of our QTL analysis may accelerate future research regarding adzuki bean traits.The development of novel markers may be useful for scientists conducting related research.
Acknowledgements
The study was supported by the National Key Research&Development Program of China (2019YFD1001300 and 2019YFD1001303),the earmarked fund for China Agriculture Research System (CARS-08) and the Agricultural Science Technology Innovation Program (ASTIP) of Chinese Academy of Agricultural Sciences.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendixassociated with this paper is available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
杂志排行

Journal of Integrative Agriculture的其它文章
- Receptor-like kinase OsASLRK regulates methylglyoxal response and content in rice
- Heredity and gene mapping of a novel white stripe leaf mutant in wheat
- Effects of temperature and solar radiation on yield of good eatingquality rice in the lower reaches of the Huai River Basin,China
- Difference in corn kernel moisture content between pre-and postharvest
- The effect of elevating temperature on the growth and development of reproductive organs and yield of summer maize
- High plant density increases seed Bt endotoxin content in Bt transgenic cotton