ZrO2改性对Ni/SBA-15催化二苯并呋喃加氢脱氧的促进作用研究
2021-06-02杜朕屹李文英
郭 良,刘 迪,杜朕屹,冯 杰,李文英
(太原理工大学省部共建煤基能源清洁高效利用国家重点实验室,山西太原030024)
热解技术是低阶煤分级分质利用的核心,煤焦油是热解技术的主要产物之一。然而,中低温煤焦油中存在大量的含氧化合物[1],这些含氧化合物降低了油品热值和稳定性,降低了油品品质,因此,必须对中低温煤焦油进行加氢精制以满足其作为燃料应用的要求[2]。中低温煤焦油中氧原子主要以酚类、呋喃类形式存在,醚、酮、羧酸、醇、酯等物质的含量相对较低[3,4]。同时各含氧化合物的加氢活性不同,相同反应条件下二苯并呋喃反应活性最低[5,6]。因而以中低温煤焦油中含量高、分子量大、反应活性低的二苯并呋喃为模型化合物可以反映催化剂的加氢脱氧性能[7−9],所得结果具有代表性。Wang等[10]研究了DBF在Pt/介孔ZSM-5分子筛、Pt/微孔ZSM-5分子筛以及Pt/γ-Al2O3催化剂上的加氢脱氧反应,发现高空速下DBF在Pt/介孔ZSM-5催化剂上脱氧程度更高,这是因为介孔ZSM-5载体有规整的介孔孔道,有利于反应物和产物的传质,同时ZSM-5载体表面较强的酸性位点也有利于脱水反应的发生,有利于含氧化合物中氧的脱除。Wang等[11]研究了DBF在SBA-15负载的贵金属催化剂上的加氢脱氧反应,发现贵金属Pt具有较好的加氢活性,Ru具有较高的脱氧产物选择性。然而,贵金属催化剂的成本相对较高,而过渡金属Ni价格低廉,适合替代贵金属催化剂进行工业化应用。加氢脱氧反应中过渡金属Ni有较高的加氢活性,但其断裂C–O键的能力相对较弱,为提高Ni基催化剂性能,研究者已展开如下研究:第一,结合其他活性金属的性质及作用,制备双金属催化剂。Dong等[12]研究了DBF在Ni、Pt单金属催化剂与NiPt双金属催化剂上的加氢脱氧反应,发现双金属催化剂的活性更高,这是因为双金属催化剂上Ni、Pt的协同作用可以促进C–O键的断裂,有利于反应的进行。第二,利用可还原金属氧化物在还原过程中部分还原产生的氧空位,来促进含氧化合物中C–O键的活化、断裂。文献[13]研究了苯酚在不同载体负载的Pd催化剂上的加氢脱氧反应,发现Pd/TiO2、Pd/ZrO2催化剂活性较好,反应结束时产物苯的选择性较高,这是因为TiO2、ZrO2载体对含氧化合物中氧原子有较强的吸附作用,从而促进了含氧化合物中氧的脱除,提高了脱氧产物的选择性。Zhao等[14]研究了顺丁烯二酸酐在Ni/ZrO2催化剂上的加氢脱氧反应,发现相对缺电子的氧空位可以吸附含氧化合物中带有孤电子的氧原子,从而活化并削弱C=O键,结合活性金属Ni良好的加氢能力,实现C=O的断裂,其中,吸附在氧空位上的氧原子与Ni上解离的氢结合并以水的形式脱除。
结合当前加氢脱氧反应中Ni基催化剂的研究进展,本研究以DBF为模型化合物,选用惰性、扩散阻力小的有序介孔SBA-15为载体,过渡金属Ni为活性金属,通过等体积浸渍方式制备了不同ZrO2含量的Ni/Zr-SBA-15催化剂及Ni/ZrO2催化剂,探究了ZrO2改性对Ni/SBA-15催化剂结构特性与反应活性的影响。
1 实验部分
1.1 催化剂的制备
实验中所用的SBA-15载体购于江苏先丰纳米材料科技有限公司。Ni/Zr-SBA-15催化剂通过分步等体积浸渍方式得到,具体制备流程为:先将一定量SBA-15载体与相应质量硝酸氧锆水溶液混合,室温搅拌24 h,90℃干燥12 h,之后在马弗炉中400℃焙烧5 h得到Zr-SBA-15载体。然后以相同步骤将过渡金属Ni浸渍在Zr-SBA-15载体上得到NiO/Zr-SBA-15催化剂前驱体,将该前驱体在管式炉中经500℃氢气气氛下还原4 h得到Ni/Zr-SBA-15催化剂。其中,催化剂上活性金属Ni的质量分数恒定为5%(质量分数),调变ZrO2在载体中的质量分数为0、2.5%、5%、10%、20%、30%,分 别 记 作Ni/SBA-15、Ni/2.5Zr-SBA-15、Ni/5Zr-SBA-15、Ni/10Zr-SBA-15、Ni/20Zr-SBA-15、Ni/30Zr-SBA-15催化剂。此外,以ZrO2为载体制备了Ni/ZrO2催化剂作为对照,其中,ZrO2载体是将硝酸氧锆在马弗炉中500℃焙烧5 h得到,之后以等体积浸渍方式将活性金属Ni负载在ZrO2载体上得到Ni/ZrO2催化剂,制备流程与Ni/Zr-SBA-15催化剂相同。
1.2 催化剂的表征
采用日本Rigaku Ultima IV型X射线衍射仪(X-ray Diffraction,XRD),以Cu靶为辐射源(λ=0.154 nm),通过小角XRD测定载体的有序结构,0.6°−5°扫描,扫描速率为0.5(°)/min;通过大角XRD测定催化剂的晶相结构,10°−80°扫描,扫描速率为4(°)/min。
采用美国Quantachrome Autosor-iQ物理吸附仪,通过Multi point BET方法分析样品的等温吸附-脱附曲线,得到样品的比表面积;通过分析吸附等温线相对压力在0.99(p/p0=0.99)下的N2吸附数据得到样品的孔容;通过对脱附支进行BJH模型分析得到样品的孔径分布。测试前将样品在300℃真空条件下预处理3 h以脱除样品表面吸附的水分及杂质,之后在−196℃下进行N2吸附-脱附实验。
采用美国麦克Autochem II-2920化学吸附仪,通过氢气程序升温还原(H2-temperature programmed reduction,H2-TPR)分析催化剂的还原特性。具体操作流程为:将0.02 g焙烧后的催化剂前驱体置于U-形石英样品管中,在Ar气氛下400°C预处理1 h,以除去样品表面吸附的水及杂质,待样品温度降至100°C时将载气切换为体积分数为10%的H2/Ar气氛,并以10 °C/min的升温速率升至900 °C进行程序升温还原实验,过程中以热导检测器(TCD)记录信号,得到H2-TPR谱图。
采用上述Autochem II-2920化学吸附仪,通过氢气程序升温脱附(H2-temperature programmed desorption,H2-TPD)分析催化剂上活性位点的数目。具体操作流程为:将0.1 g催化剂置于U-形石英样品管中,在体积分数为10%的H2/Ar气氛下500°C预处理2 h,Ar吹扫30 min,待样品温度降至50°C时再次以10%的H2/Ar处理2 h,Ar吹扫2 h,之后样品以10℃/min升温速率升至400℃进行氢气程序升温脱附实验,脱附过程中TCD检测记录信号,得到H2-TPD谱图。同时根据催化剂H2-TPD分析结果计算了催化剂上活性金属Ni的分散度,计算公式如(1)所示,其中,nH2为H2-TPD中脱附氢气物质的量,nNi为催化剂中活性金属Ni物质的量,SF为化学计量因子(Ni/H),SF= 2。

采用上述Autochem II-2920化学吸附仪,通过氧脉冲分析催化剂上氧空位的相对含量。具体操作流程如下:将0.02 g催化剂置于U-形石英样品管中,在体积分数为10%的H2/Ar气氛下500℃预处理2 h,He吹扫30 min并开始降温,待样品温度降至340℃时切换为体积分数3%的O2/He气氛进行氧脉冲实验,脉冲间隔为3 min,通过TCD检测记录信号,直至脉冲信号峰强度不发生变化时结束实验。通过计算催化剂在脉冲过程中氧气的吸附量,得到催化剂中氧空位的相对含量。
采用吡啶原位吸附红外光谱测定催化剂上Brønsted、Lewis酸量,设置红外光谱仪(德国Bruker,TENSOR 27)分 辨 率 为4 cm−1,谱 图 累 加32次,谱图采集为600–4000 cm−1。其中,1540 cm−1处吸收峰为Brønsted酸位点特征峰,1450 cm−1处吸收峰为Lewis酸位点特征峰。同时测定了不同脱附温度下酸性位点的特征峰,以反映催化剂酸强度的变化,其中,脱附温度为150℃时,得到催化剂的总酸量,脱附温度为300℃时,得到催化剂上中强酸酸量。
采用日本岛津AXIS Supra的X射线光电子能谱(X-ray photoelectron spectroscopy,XPS),得到催化剂中氧空位及表面原子的相关信息。XPS的激发源为单色Al靶,在谱图分析前以污染碳C 1s峰结合能284.8 eV对谱图进行荷电校正。
1.3 催化剂性能评价
在高压反应釜中对催化剂加氢脱氧性能进行评价。反应原料由3.0%的DBF、1.0%正十二烷和96.0%正癸烷组成,催化剂质量为60 mg,反应温度为280℃,室温下初始氢气压力为4 MPa,加热至280℃时氢气压力升至6.5 MPa,反应180 min,过程中每10 min从取样管在线取样,并通过气相色谱-质谱-氢火焰离子化检测器联用仪(GC-MSFID)对产物组成进行定性(MS)和定量(FID),色谱柱为HP-5ms(30 m×0.25 mm×0.25μm)。以正十二烷为内标通过内标法对产物组成进行定量,并计算反应过程中催化剂上DBF转化率(xDBF)、产物选择性(si)、产物收率(yi)、碳平衡、DBF反应速率(rDBF)及目标产物BCHs生成速率(rBCHs),具体计算公式如下所示:

式中,n0为反应初始时DBF物质的量(mol);n为 反应过程中DBF物质的量(mol);ni为反应过程中某产物i的物质的量(mol);t为反应时间(min);mcat为催化剂质量(g)。为了对比催化剂本征反应活性,在低转化率下计算了DBF本征反应速率、目标产物BCHs,包含联环己烷(Bicyclohexyl,BCH)及其同分异构体环戊基甲基环己烷(Cyclopentylmethylcyclohexane,CPMCH)的生成速率及产物选择性。在计算DBF反应速率时t取10 min,在计算BCHs生成速率及产物选择性时t取20 min。
2 结果与讨论
2.1 催化剂的表征
2.1.1 有序介孔结构及晶相结构
图1 为SBA-15与ZrO2改性SBA-15的小角XRD谱图。由图1可以看到,SBA-15、2.5Zr-SBA-15、5Zr-SBA-15载体在2θ为0.8°、1.4°、1.6°处有明显的衍射峰,分别对应载体的(100)、(110)、(200)特征峰,表明这些载体均有规整的孔道,且孔道符合二维六方结构[15],同时也说明适量ZrO2的添加(ZrO2含量低于5%,质量分数)仍然可以使SBA-15载体的有序介孔结构得以保持,而当ZrO2含量大于5%时,可以看到载体的小角XRD衍射峰逐渐减弱并消失,表明过量ZrO2的添加填充到了SBA-15孔道当中,降低了载体的长程有序度[16]。对于ZrO2载体,可以看到其小角XRD谱图为一条平滑曲线,无衍射峰,表明ZrO2载体没有有序介孔结构。
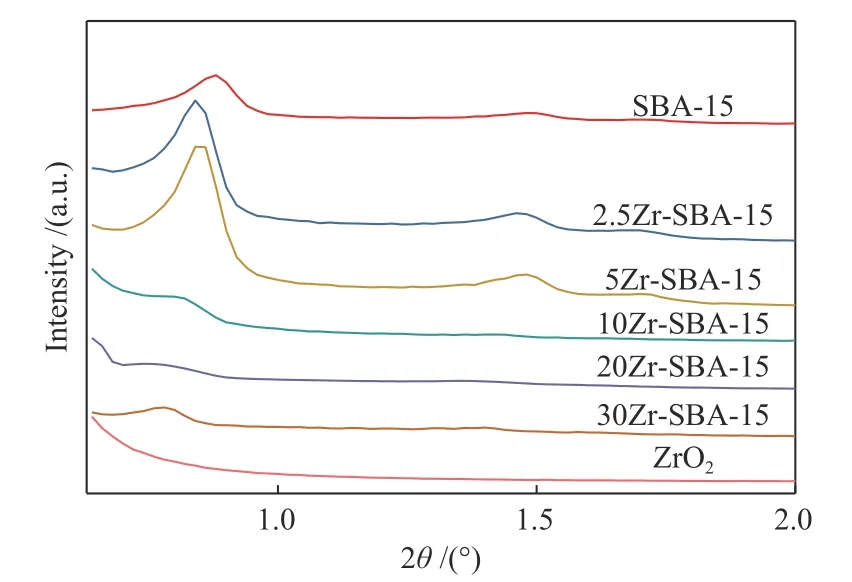
图1 SBA-15载体与Zr-SBA-15载体的小角XRD谱图Figure 1 Small-angle XRD patterns of SBA-15 and Zr-SBA-15 supports
图2 为还原态Ni/Zr-SBA-15催化剂的XRD谱图,图2中“t-ZrO2”和“m-ZrO2”分别表示ZrO2的四方相和单斜相。由图2可以看到,对于Ni/Zr-SBA-15催化剂,其在2θ为44.5°、51.8°和76.4°附近均有衍射峰,分别对应Ni的(111)、(200)和(220)晶面[17];对于未ZrO2改性Ni/SBA-15催化剂,其仅有Ni的衍射峰。随着ZrO2的添加,催化剂在2θ为30.1°、50.2°和59.7°附近出现衍射峰,为t-ZrO2(四方相),且随着ZrO2的增加,衍射峰强度增强,这是因为当ZrO2含量较低时(低于10%),ZrO2分散度高,衍射峰强度较低,随着ZrO2的进一步增加(高于10%),ZrO2开始富集,衍射峰强度增强。对于Ni/ZrO2催化剂,除存在t-ZrO2(四方相)外,其在2θ为24.4°、28.2°和31.4°附近也出现衍射峰,为m-ZrO2(单斜相),且以m-ZrO2为主,因此,Ni/ZrO2催化剂会表现出与Ni/Zr-SBA-15催化剂不同的衍射峰。
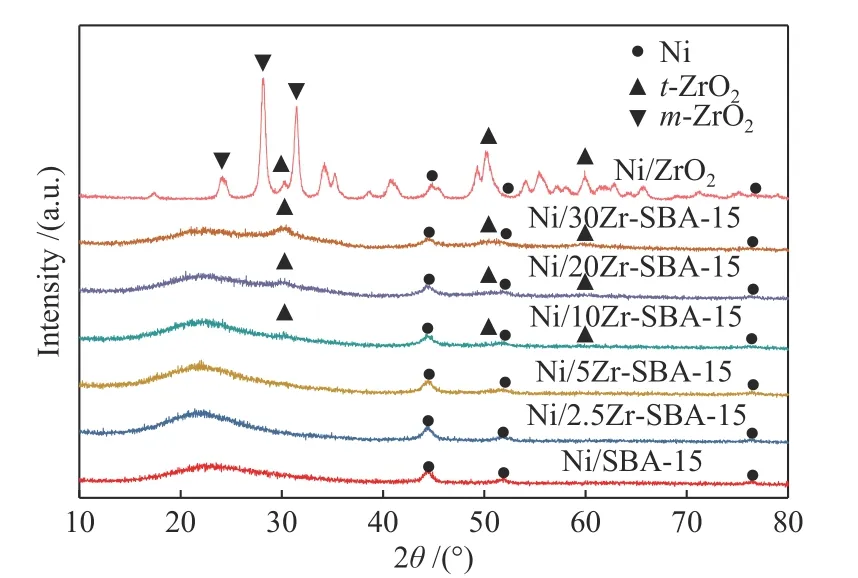
图2 还原态催化剂的XRD谱图Figure 2 XRD patterns of reduced catalysts
同时根据谢乐公式,计算了Ni/Zr-SBA-15催化剂上Ni晶粒尺寸,如表1所示。由表1可以看到,Ni/SBA-15催化剂上Ni晶粒尺寸为9.7 nm,随着ZrO2的增加Ni晶粒尺寸逐渐减小,当ZrO2含量为2.5%、5%、10%、20%、30%时,Ni晶粒尺寸分别为8.2、8.0、7.5、7.3、6.7,表明ZrO2的添加减小了Ni的晶粒尺寸,促进了Ni的分散。对于Ni/ZrO2催化剂,其Ni晶粒尺寸为6.8 nm,与Ni/30Zr-SBA-15催化剂晶粒尺寸相近。

表1 催化剂的化学性质Table 1 Chemical properties of catalysts
2.1.2 比表面积及孔径分布
图3 (a)、(b)分别为Ni/Zr-SBA-15催化剂的氮气物理吸附-脱附曲线及孔径分布。可以看到,该催化剂的氮气物理吸附-脱附等温线为IV型吸附-脱附等温线,且有明显的H1型滞后环,表明该催化剂为有序介孔结构[18]。与其他催化剂相比,Ni/ZrO2催化剂的吸附-脱附等温线基本重合,不是IV型吸附-脱附等温线,无H1型滞后环,表明Ni/ZrO2催化剂没有有序介孔结构,这与小角XRD的表征结果相吻合。
根据上述催化剂氮气物理吸附-脱附曲线及孔径分布图,表2给出了Ni/Zr-SBA-15催化剂比表面积、孔容及最可几孔径。可以看到,Ni/SBA-15催化剂的比表面积为493.6 m2/g,孔容为1.11 cm3/g,最可几孔径为7.8 nm。随着ZrO2的增加,Ni/Zr-SBA-15催化剂的比表面积、孔容逐渐减小,表明ZrO2的添加会堵塞催化剂孔道。当ZrO2添加量为30%时,比表面积降至360.0 m2/g,孔容降至0.77 cm3/g,最可几孔径为7.8 nm。同时可以看到,Ni/ZrO2催化剂具有较小的比表面积、孔容,两者分别为46.9 m2/g和0.13 cm3/g。
2.1.3 催化剂的酸性
图4 为Ni/Zr-SBA-15催化剂的吡啶吸附红外光谱谱图,其中,1450 cm−1处为Lewis酸的特征峰,1540 cm−1处为Brønsted酸的特征峰[19],可以看到,Ni/SBA-15催化剂在1450 cm−1处吸附峰强度较弱,在1540 cm−1处无吸附峰,表明Ni/SBA-15催化剂有少量的Lewis酸,而没有Brønsted酸。与Ni/SBA-15催化剂相比,可以看到,随着ZrO2的增加,Ni/Zr-SBA-15催化剂在1450 cm−1处吸收峰呈现出先增大后减小的趋势,1540 cm−1处仍没有吸收峰,表明ZrO2的添加仅在催化剂中引入了Lewis酸,而没有Brønsted酸,且随着ZrO2的增加Lewis酸量表现出先增加后减少的趋势。

图4 催化剂的吡啶红外光谱谱图Figure 4 Py-FTIR profiles of catalysts
表3 列出了催化剂中Lewis酸的酸量。由表3可以看到,Ni/SBA-15催化剂的总酸量为8.2 μmol/g,中强酸酸量为3.8μmol/g,催化剂酸性较弱。随着ZrO2的添加,催化剂酸量增加,这是因为ZrO2中含配位不饱和的Zr3+及Zr4+,其作为Lewis酸位点,有效增强了催化剂酸性。在ZrO2负载量高于10%(质量分数)时,催化剂酸量下降,这是因为过量的ZrO2降低了催化剂比表面积、孔容,从而降低了催化剂酸量。可以看到,Ni/10Zr-SBA-15催化剂的酸量最高,总酸量为84.5μmol/g,中强酸酸量为30.5μmol/g。同时也分析了催化剂中中强酸在总酸量中的占比,可以看到Ni/SBA-5催化剂上该占比为0.46,随着ZrO2的增加,中强酸在总酸量中的占比不断减小,当ZrO2添加量为30%时,该占比下降至0.20。以上结果表明,ZrO2改性在催化剂中引入了Lewis酸,且引入的Lewis酸酸性位点以弱酸为主。

表3 催化剂的酸量Table 3 The acid amount of catalysts
2.1.4 氧空位浓度
图5 (a)为Ni/Zr-SBA-15催化剂的H2-TPR谱图。可以看到,ZrO2载体在较高温度下有氢气消耗峰,表明ZrO2被部分还原,产生了氧空位。对于Ni/Zr-SBA-15催化剂,可以发现随着ZrO2的增加,氢气消耗峰逐渐向高温方向移动,表明ZrO2的添加增强了Ni物种与载体间相互作用,H2-TPR实验规律较好。同时结合表1可以发现,ZrO2含量不同催化剂上Ni分散度不同,这也会影响催化剂的还原性能。一方面ZrO2的添加增强了Ni物种与载体间相互作用,提高了Ni物种的还原温度;另一方面随着ZrO2的添加,Ni分散度增加,Ni晶粒尺寸减小,Ni物种还原温度升高,从而也会影响催化剂的还原性能。
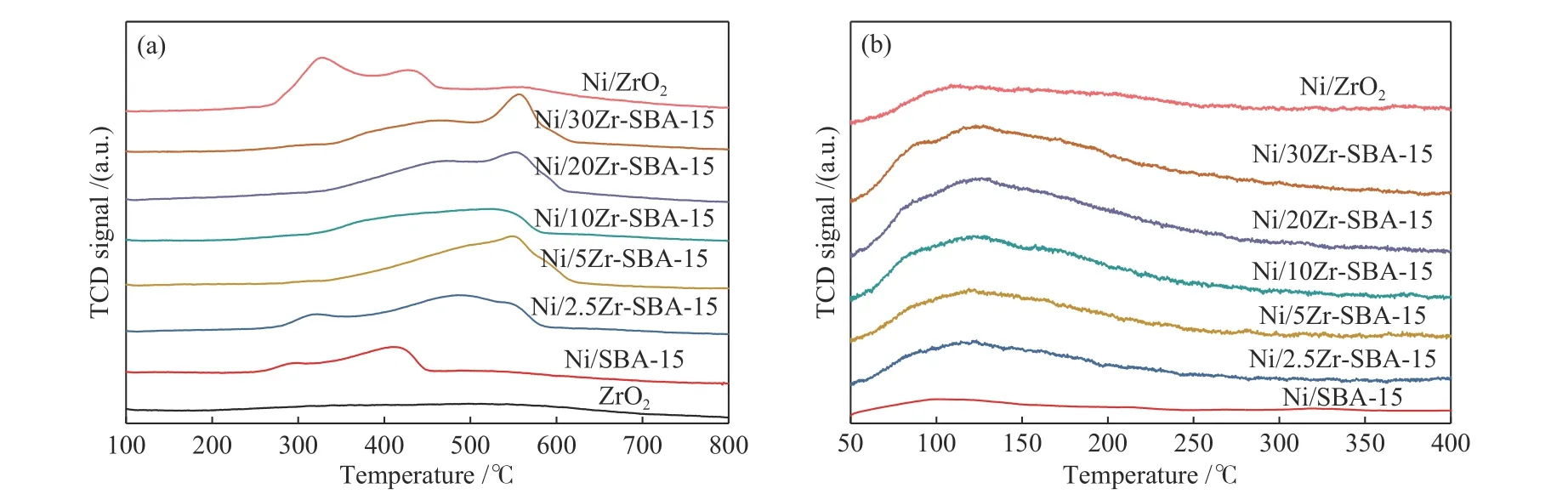
图5 催化剂的H2-TPR (a)及H2-TPD(b)谱图Figure 5 H2-TPR (a)and H2-TPD(b)profiles of catalysts
同时分析了催化剂在还原过程中氢气的消耗量,结果如表1所示。可以看到,随着ZrO2的添加,氢气消耗量增加,表明在催化剂前驱体的还原过程中,除Ni物种的还原外,还涉及ZrO2的部分还原,且随着ZrO2的增加,催化剂中氧空位浓度增加。对于Ni/ZrO2催化剂,其H2-TPR谱图与Ni/Zr-SBA-15催化剂谱图相比存在明显差异,根据本文现有数据推测,这是因为Ni/ZrO2催化剂中ZrO2主要以单斜相形式存在,而Ni/Zr-SBA-15催化剂中ZrO2主要以四方相形式存在,四方相ZrO2的存在提高了Ni物种与载体间相互作用,提高了Ni物种的还原温度,与文献报道一致[20,21],因此,Ni/ZrO2催化剂与Ni/Zr-SBA-15催化剂表现出不同的还原温度。
图5 (b)为Ni/Zr-SBA-15催化剂的H2-TPD谱图。可以看到,Ni/Zr-SBA-15催化剂在125℃附近有氢气脱附峰,且随着ZrO2的添加,脱附峰逐渐变大,表明Ni/Zr-SBA-15催化剂上有更多的氢活性位点。同时表1根据催化剂H2-TPD的表征结果,分析了催化剂上活性金属Ni的分散度,可以看到随着ZrO2的增加,H2脱附量增加,催化剂上活性金属Ni分散度提高,这与XRD上Ni晶粒尺寸的变化趋势相吻合。而对于Ni/ZrO2催化剂,其活性金属Ni的分散度仅为6.6%,这是由于ZrO2载体的比表面积较小,不利于活性金属Ni的分散造成的。
通过O2-Pulse实验,分析了催化剂中氧空位的相对含量,结果如表1所示。可以看到,Ni/SBA-15催化剂的氧气消耗量为206.7μmol/g,由于SBA-15载体是不可还原氧化物,故该催化剂上氧气消耗归因于活性金属Ni的氧化消耗。对于Ni/Zr-SBA-15催化剂,其Ni负载量均为5%,可以看到随着ZrO2的增加,催化剂的氧气消耗量增加,均大于206.7μmol/g(Ni/SBA-15),表明Ni/Zr-SBA-15催化剂上除活性金属Ni对氧气的消耗外,还伴随着氧空位对氧气的吸附消耗,这与上述H2-TPR的分析结果相吻合。
图6 (a)为Ni/Zr-SBA-15催化剂Zr 3d轨道的能谱图,通过双峰拟合将该谱图拟合为四个峰,峰位置分别为181.7、182.5、184.1、184.8 eV,其中,181.7、184.1 eV处的峰归属于Zr3+,182.5、184.8 eV处的峰归属于Zr4+,谱图中Zr3+的存在表明ZrO2被部分还原,有氧空位生成[22−24]。对于Ni/Zr-SBA-15催化剂,其O 1s轨道能谱图与Ni/SBA-5催化剂O 1s轨道能谱图均只有一个特征峰,为SiO2载体Si−O−Si键的特征峰,表明SBA-15载体中Si−O−Si键的信号太强,掩盖了ZrO2中氧原子的能谱信息。因此,为了更加鲜明地区分氧空位与晶格氧特征峰,选用代表性强的Ni/SBA-15催化剂及Ni/ZrO2催化剂进行O 1s轨道特征峰分析。图6(b)给出了Ni/SBA-15、Ni/ZrO2催化剂O 1s轨道的能谱图,可以看到两者O 1s轨道的能谱图存在明显差别,对于Ni/SBA-15催化剂,其O 1s轨道的能谱图有一个明显的特征峰,电子结合能为532.8 eV,该峰归属于SBA-15载体中氧的特征峰[25,26]。对于Ni/ZrO2催化剂,其O 1s轨道的能谱图有两个特征峰,电子结合能分别为529.7、531.4 eV,分别归属于ZrO2中 的晶格 氧与氧 空位[23,24, 27,28],这 表 明ZrO2的添加可以在催化剂中引入氧空位。

图6 Ni/Zr-SBA-15催化剂Zr 3d轨道(a)及Ni/SBA-15、Ni/ZrO2催化剂O 1s轨道能谱图(b)Figure 6 XPS profiles of Ni/Zr-SBA-15 catalysts:Zr 3d (a)and Ni/SBA-15, Ni/ZrO2 catalysts:O 1s(b)
2.2 反应性能评价
图7 为未改性Ni/SBA-15催化剂上DBF转化率及产物收率随反应时间的变化,反应过程中碳平衡均在95%−105%,可以看到反应100 min时DBF完全转化,反应过程中同时伴随中间产物的生成及转化。

图7 二苯并呋喃在Ni/SBA-15催化剂上转化率(a)及产物收率(b)随时间的变化Figure 7 DBF conversion (a)and product yields(b)over Ni/SBA-15 catalysts as a function of time
从图7(b)中可以看到,DBF转化生成四氢二苯并呋喃(THDBF)、六氢二苯并呋喃(HHDBF)、十二氢二苯并呋喃(DHDBF)、2-环己基苯酚(CHPOH)、2-环己基环己醇(CHCHOH)、环己基环己酮(CHCHO)、环己基苯(CHB)、环己基环己烯(CHCHE)、BCHs等物质,其中,THDBF、HHDBF、CHPOH、CHCHO、CHB、CHCHE等物质在反应结束时均完全转化,而DHDBF、CHCHOH的转化相对较慢,使得反应结束时DHDBF、CHCHOH、BCHs成为主要的三种反应产物。
根据本实验及前期DFT计算结果[29,30],作者总结了DBF在Ni催化剂上加氢脱氧的反应网络,如图8所示。DBF先加氢生成THDBF、HHDBF,生成的THDBF、HHDBF同时会发生开环反应和加氢反应,高压下倾向于发生加氢反应生成DHDBF,之后开环生成CHCHOH,同时由THDBF、HHDBF开环生成的CHPOH在高压下也倾向于发生加氢反应生成CHCHOH,之后CHCHOH再脱羟基生成目标产物BCHs;同时HHDBF也可以通过开环反应再加氢形成环己基环己酮CHCHO,之后CHCHO加氢脱羟基生成BCHs。在DBF加氢脱氧反应网络中DHDBF发生开环反应的活化能最高,同时CHCHOH在活性金属Ni表面的吸附能较小,脱氧活化能较高,反应速率较慢,因此,在反应结束时DHDBF、CHCHOH、BCHs有较高的产物收率。
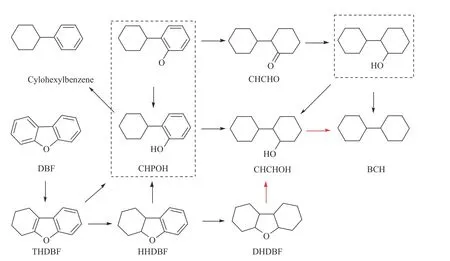
图8 二苯并呋喃在Ni基催化剂上加氢脱氧反应路径图Figure 8 Reaction pathways for HDO of DBF over Ni-based catalysts
在上述Ni/SBA-15催化剂性能评价中,可以发现未改性Ni/SBA-15催化剂断裂C–O键能力不足,反应结束时含氧中间产物DHDBF和CHCHOH均有较高的收率。通过对催化剂进行ZrO2改性,降低含氧中间产物C–O键断裂能垒,以促进反应的进行,为此对改性的Ni/Zr-SBA-15催化剂进行反应评价,并以主要产物DHDBF、CHCHOH、BCHs收率来衡量催化剂性能。
图9 (a)为Ni/SBA-15、Ni/10Zr-SBA-15、Ni/ZrO2催化剂上DBF转化率随反应时间的变化,反应过程中碳平衡均在95%−105%。可以看到,反应结束时三种催化剂上DBF转化率均为100%,而在反应初始时DBF转化率存在明显差异,反应10 min时低转化率条件下DBF反应速率从大到小依次为Ni/10Zr-SBA-15> Ni/ZrO2> Ni/SBA-15,表 明ZrO2的添加加速了反应物DBF的转化,促进了反应的进行。
图9 (b)为DBF加氢脱氧反应过程中Ni/SBA-15、Ni/10Zr-SBA-15、Ni/ZrO2催化剂上主要产物收率随反应时间的变化。Ni/SBA-15催化剂上目标产物BCHs的收率仅为32%,反应结束时DHDBF、CHCHOH的收率分别高达40%、24%,说明无ZrO2改性的Ni/SBA-5催化剂上金属Ni本身对含氧中间产物的转化较慢。在添加10%的ZrO2后,可以看到Ni/10Zr-SBA-15催化剂上主要产物为目标产物BCHs,而含氧中间产物DHDBF、CHCHOH在反应结束后已完全转化。在反应20 min时两者的收率达最大值,分别为19%、5.4%,之后快速转化,这表明ZrO2的添加明显加快了含氧中间产物的进一步加氢脱氧,提高了目标产物BCHs的收率。对于Ni/ZrO2催化剂,可以看到反应结束时含氧中间产物DHDBF的收率较低,仅为2.7%,含氧中间产物CHCHOH的收率较高,为41%,目标产物BCHs收率为48%,催化活性明显低于Ni/10Zr-SBA-15催化剂。

图9 Ni/SBA-15、Ni/10Zr-SBA-15、Ni/ZrO2催化剂上DBF转化率(a)及主要产物收率(b)随时间的变化Figure 9 DBF conversion rate (a)and product yields (b)over Ni/SBA-15, Ni/10Zr-SBA-15, Ni/ZrO2 catalysts as a function of time
图10 为DBF反应速率及目标产物BCHs生成速率随ZrO2含量的变化。可以看到,随着ZrO2的增加,两者均表现出先增加后减小的趋势。对于未ZrO2改性的Ni/SBA-15催化剂,其DBF反应速率较小,仅为1.25 mmol/(min·g);随着ZrO2含量的增加,DBF反应速率增加,当ZrO2含量为10%时,DBF反应速率达到最大值,为9.21 mmol/(min·g);当ZrO2添加量大于10%时,DBF反应速率开始下降。同时对比不同催化剂上目标产物BCHs的生成速率,可以发现当ZrO2添加量为20%时BCHs生成速率最大,为3.74 mmol/(min·g),这说明Ni位点与ZrO2之间的协同作用存在最佳匹配,即Ni位点上主要发生H2的吸附解离、DBF及加氢中间产物的吸附和加氢反应,而ZrO2除了促进Ni颗粒的分散外,其提供的氧空位可以促进含氧中间产物上C–O键的断裂。因此,DBF转化速率主要由暴露的Ni位点决定,而BCHs的生成速率是Ni位点和ZrO2之间的协同匹配决定,从而使得Ni/20Zr-SBA-15催化剂相较Ni/10Zr-SBA-15催化剂DBF反应速率更低,但BCHs生成速率却更高。DBF反应速率及目标产物BCHs生成速率先增加是因为ZrO2的添加减小了活性金属Ni的晶粒尺寸,促进了Ni颗粒的分散,同时提供了氧空位,从而加速了反应的进行;当ZrO2含量为30%时DBF反应速率减小,是因为过量ZrO2的添加降低了催化剂的比表面积,同时由于金属和载体间强相互作用,使得活性金属Ni周围部分还原的ZrOx会迁移并覆盖Ni活性位点[31,32],从而不利于DBF的转化。
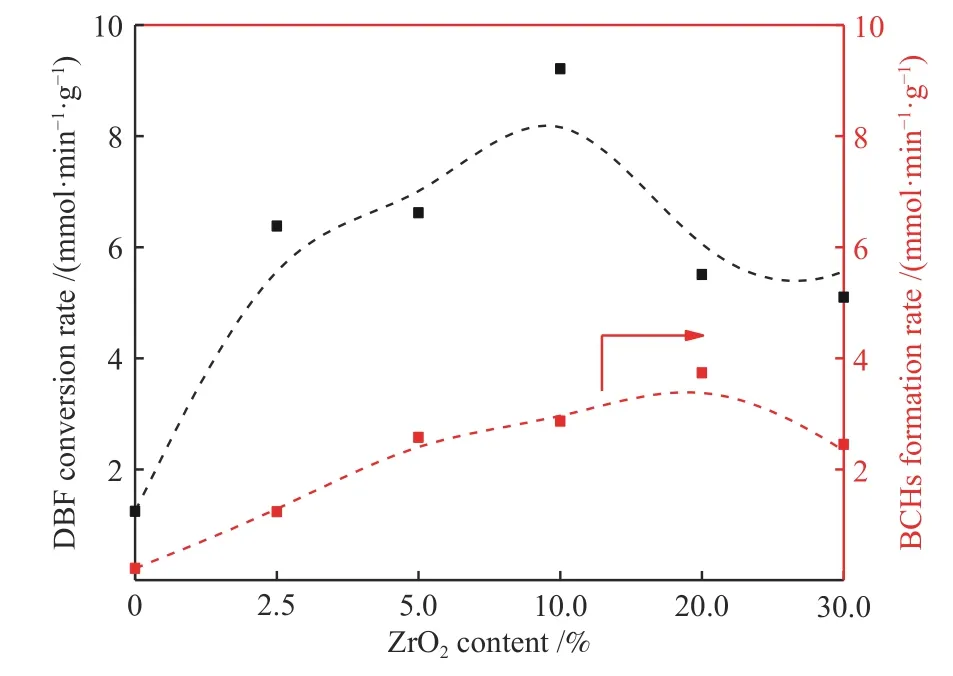
图10 Ni/Zr-SBA-15催化剂上DBF反应速率、BCHs生成速率随ZrO2含量的变化Figure 10 DBF reaction rate and BCHs formation rate over Ni/Zr-SBA-15 catalysts
加氢脱氧反应中催化剂酸性对反应活性也有很大的影响,催化剂酸性可以协同活性相促进含氧中间产物发生开环反应以及促进醇脱水反应的发生。在Ni/Zr-SBA-15催化剂酸性分析中,可以发现Ni/Zr-SBA-15催化剂上仅有Lewis酸,且Lewis酸量随ZrO2的增加表现出先增大后减小的趋势,Ni/10Zr-SBA-15催化剂上Lewis酸酸量最大。催化剂上的Lewis酸包括配位不饱和的金属阳离子和氧空位,氧空位能够吸附含氧化合物,促进含氧化合物中C–O的活化、断裂[14,33]。因此,分析了Ni的加氢性能(金属分散度)和氧空位对于产物选择性的关联,以进一步确认Ni金属分散度和氧空位在反应过程中的作用机理。
图11 为主要产物DHDBF、CHCHOH、BCHs及部分中间产物选择性与ZrO2含量、Ni分散度及氧空位浓度变化的关系图。可以看到,对于THDBF、HHDBF、CHPOH,其选择性在Ni/SBA-15催化剂上最高,之后随着ZrO2的添加催化剂上Ni分散度提高、氧空位浓度增加,这几种产物的选择性随Ni分散度、氧空位浓度的升高而逐渐降低,表明THDBF、HHDBF、CHPOH的转化被促进。对于DHDBF、CHCHOH,可以看到其选择性随ZrO2含量、Ni分散度、氧空位浓度表现出先增加后减小的趋势,选择性在Ni/2.5Zr-SBA-15催化剂上最高,这是因为当ZrO2少量添加时,由于Ni分散度的提高,更多的Ni活性位点可以将DBF、THDBF、HHDBF(初级中间产物)快速转化为DHDBF、CHCHOH等次级中间产物;但如前所述,DHDBF、CHCHOH的进一步加氢脱氧反应困难、能垒较高,故少量ZrO2的添加提供的氧空位数量有限,不足以将其迅速进一步转化为BCHs,故DHDBF、CHCHOH发生累积而体现为Ni/SBA-15上两者的选择性更高。随着ZrO2的进一步添加,催化剂断裂C–O键能力增强,DHDBF、CHCHOH的转化速率加快,选择性降低。但是,当ZrO2含量为30%时,过量ZrO2的Ni活性位点的覆盖效应明显,导致中间产物选择性又有上升,同时BCHs选择性下降。以上结果进一步证实了高分散的Ni颗粒可以与氧空位发生协同作用促进含氧中间产物的转化,提高目标产物的收率。

图 11 Ni/Zr-SBA-15 催化剂上产物选择性与 ZrO2 含量、Ni 分散度、氧空位浓度的变化Figure 11 The selectivity of products onNi/Zr-SBA-15catalysts varies with Zr O2 content, Ni dispersion, and oxygen vacancyconcentration
3 结 论
ZrO2的添加会提高Ni颗粒分散度、引入氧空位,进而影响加氢脱氧反应的性能。随着ZrO2的添加,Ni/SBA-15催化剂活性表现出先上升后下降的趋势,当ZrO2添加量为10%时,二苯并呋喃反应速率最高,为9.21 mmol/(min·g);当ZrO2添加量为20%时联环己烷生成速率最高,为3.74 mmol/(min·g)。这是因为适量ZrO2的添加增强了Ni与载体间相互作用,促进了Ni颗粒的分散,有利于DBF的转化,同时ZrO2的添加也在催化剂中引入了氧空位,促进了含氧化合物中C−O键的断裂,有利于加氢脱氧反应的进行;而过量ZrO2的添加降低了催化剂的比表面积,不利于Ni颗粒的分散,不利于反应的进行。
