基于宏基因组学的新疆牧场牛潜在人兽共患病原菌调查
2021-06-01
(1.南华大学公共卫生学院,湖南省衡阳市421001;2.中国疾病预防控制中心传染病预防控制所国家重点实验室,北京市102206;3.石河子大学动物科技学院,新疆维吾尔自治区石河子市832003;4.新疆生产建设兵团第八师石河子市动物卫生监督所,新疆维吾尔自治区石河子市832000)
在全球范围内,对人类和动物宿主的健康具有潜在影响的病原体被频繁地检测到[1]。大多数导致人类疾病的新发的病原体都是人兽共患病原菌,人兽共患病可以从野生或者家养动物传染给人类,对公共卫生和人类健康造成了重大威胁[2-4]。新疆是中国最大畜牧业生产基地之一,与8个国家接壤,国际性贸易频繁[5]。近些年畜牧业不断地发展,致病菌在人群中传播速度加快,从而动物源人兽共患病的流行风险也随之明显增加。牛作为新疆地区畜牧业的主要动物之一,与人们的生活生产密切相关,是人们重要的蛋白质来源[6]。因此,了解规模化养殖牛的潜在病原菌携带状况调查很有必要,对有效防治人兽共患病具有重要意义。在牧场的生产实践中,传统细菌病原的检测耗时长,检测量有限,难以满足需求[7]。宏基因组是特定生物环境中全部微生物遗传物质的总和[8]。宏基因组学二代测序技术直接获取样本中所有核酸片段的序列信息,与特定的数据库比对结合生物信息分析,检测出所有微生物的种类及序列数量[9],目前已成为国际微生物学研究的热点,成为开发新的生物活性物质、研究群落中微生物多样性的新途径,而且宏基因组学技术自动化程度高,可以准确灵敏地鉴定病原微生物[10]。本研究基于二代宏基因组学技术对新疆规模化养殖牧场牛群的鼻咽和肛拭子样品中细菌种类进行分析鉴定,探讨牛携带潜在病原以及人兽共患病主要菌种状况,较系统地评价潜在病原菌对宿主自身和人类健康的影响,为相关疾病的防控提供基础科学依据。
1 材料和方法
1.1 样品采集
2019年8月从新疆南疆和北疆集中大牧场的牛群中采集牛鼻咽拭子303份和肛拭子306份,其中南疆喀什市采集牛鼻咽拭子和牛肛拭子各104份,北疆石河子市采集牛鼻咽拭子199份和牛肛拭子202份。采集过程中均使用一次性用具以避免样品交叉污染,所有的样品经无菌容器密封好后放入车载冰箱运回实验室,样品保存在-70 ℃超低温冰箱。
1.2 宏基因组DNA的提取及BGISEQ测序
将待测样品完全解冻后,采用QIAamp DNA micro Kit提取基因组DNA。检测DNA纯度和降解程度后,去除不合格样品,将合格的鼻咽拭子/肛拭子DNA样品,分别根据样品编号顺序按质量1∶1比例每10份混合为一组,即鼻咽拭子和肛拭子各自混合为鼻咽拭子组和肛拭子组,不交叉混合。对混合后的DNA样品进行超声片段化后,依据BGISEQ进行DNA文库构建,宏基因组测序由华大基因科技有限公司完成。
1.3 数据分析
对于原始测序数据采用Trimmomatic去除低质量测序数据;使用Kraken2以及配套的Minikrakenv2数据库进行物种分类;基于物种丰度信息表,对不同类别样品以及不同地区的样本分别应用R语言进行基于Bray、Euclidean距离的主坐标分析(principal coordinate analysis,PCoA)、相似性分析(analysis of similarities,Anosim)和多元方差分析(Adonis)。应用Python进行Wilcoxon秩和检验分析潜在人畜共患病原菌的差异性。
2 结 果
2.1 测序数据
BGISEQ测序原始数据通过质控排除一定比例低质量数据,获得高质量序列数据。对于一条序列,每10个碱基为一个窗口计算碱基平均测序质量,如果低于20,则在该窗口处切断,并且只保留双端都大于121 bp的reads。经数据质控所得29组肛拭子样品和22组鼻咽拭子样品的宏基因组数据进行分析。
2.2 微生物群落组成
根据宏基因组序列分析,共得到4个界40个门81个纲175个目398个科1 283个属和4 218个种。在界分类水平上,所有样品中相对丰度最大的均为细菌。其中,29组肛拭子样品相对丰度为86.90%~98.42%;22组鼻咽拭子样品相对丰度为74.51%~92.91%。这说明新疆牧场牛鼻咽部和大肠直肠部位的微生物群落组成主要是细菌。
2.3 不同样品的细菌种类分析
2.3.1 不同类别牛样品的细菌种类注释分析 基于物种注释结果,只保留在至少1个样本中丰度大于0.01%的菌种,分析牛样品在门和属水平上相对丰度排名前10的细菌种类(图1)。由图1可知,在门分类水平上,肛拭子和鼻咽拭子样品中细菌物种占主导地位的门为放线菌门(Actinobacteria)、变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)和拟杆菌门(Bacteroidetes)。在属分类水平上,肛拭子样品中优势菌属为双歧杆菌属(Bifidobacterium)、异壁放线菌菌属(Actinoalloteichus)、埃希氏菌属(Escherichia)和芽孢杆菌属(Bacillus);而鼻咽拭子样品中优势菌属为异壁放线菌菌属、芽孢杆菌属、不动杆菌属(Acinetobacter)和丛毛单胞菌属(Comamonas)。
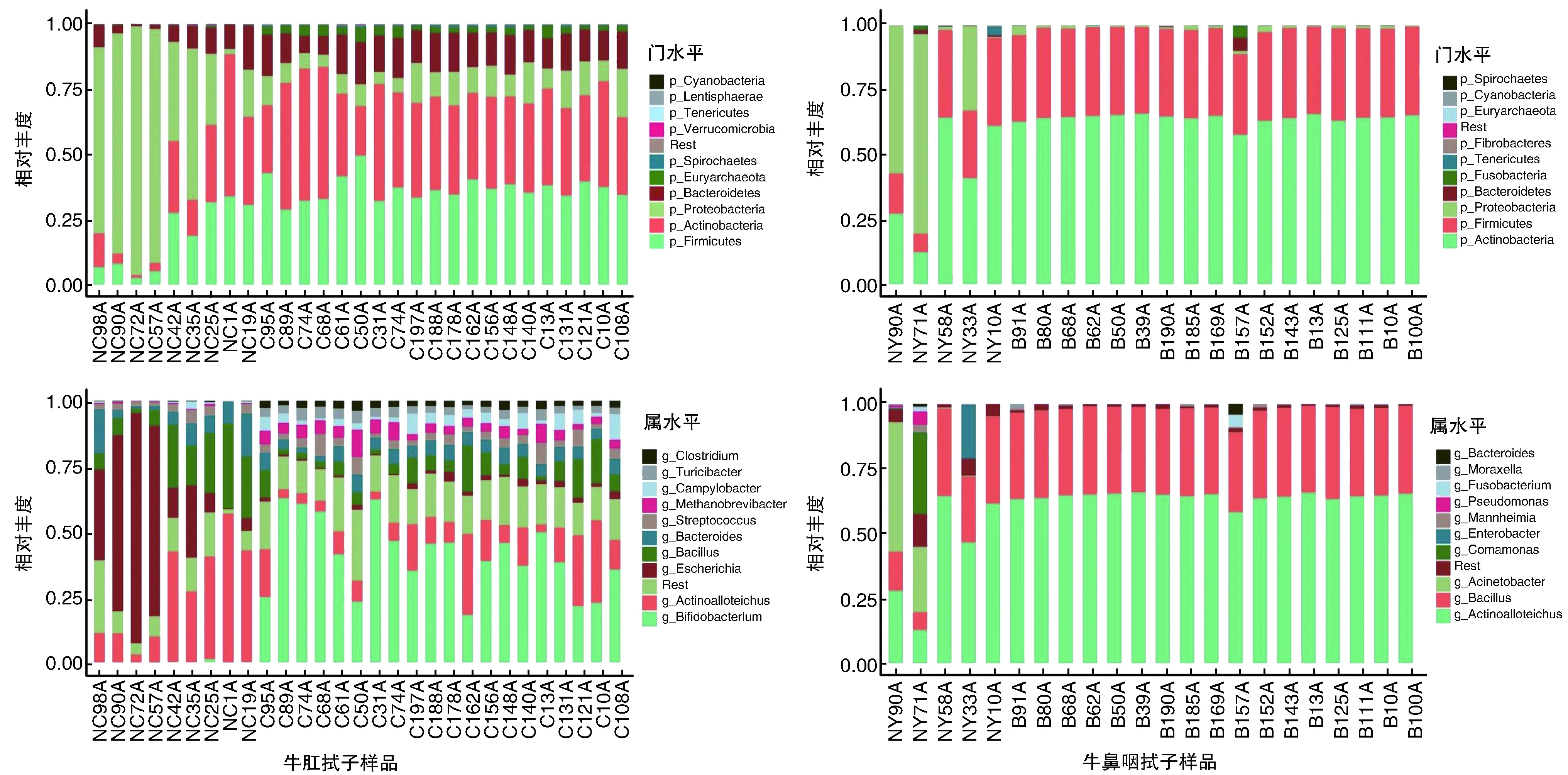
图1 不同类别牛样品中细菌种类排名前10位的物种丰度柱状图
2.3.2 不同类别牛样品细菌群落的比较 通过PCoA分析不同样品细菌菌群的β多样性(图2)。基于不同样品类别,在门水平上,第一主成分(PC1)和第二主成分(PC2)对所有样品差异的贡献率分别为87.32%和11.94%,累积贡献率为99.26%(图2A);在属水平上,PC1和PC2对所有样品差异的贡献率分别为66.85%和19.47%,累积贡献率为86.32%(图2B)。无论在门水平还是属水平,两组样品在PCoA图上均能独自聚类,但有少部分重叠。基于不同地区分布,喀什市和石河子市的肛拭子样品组出现交叠现象(图2C),而基于属分类水平两者发生了明显的彼此分离现象,表现为独立的分布(图2D)。在不同分级(门和属)水平上(图2E和2F),石河子市鼻咽拭子样品基本处于中心点左侧,而喀什市鼻咽拭子样品组在中心点的左侧和右侧都有分布。
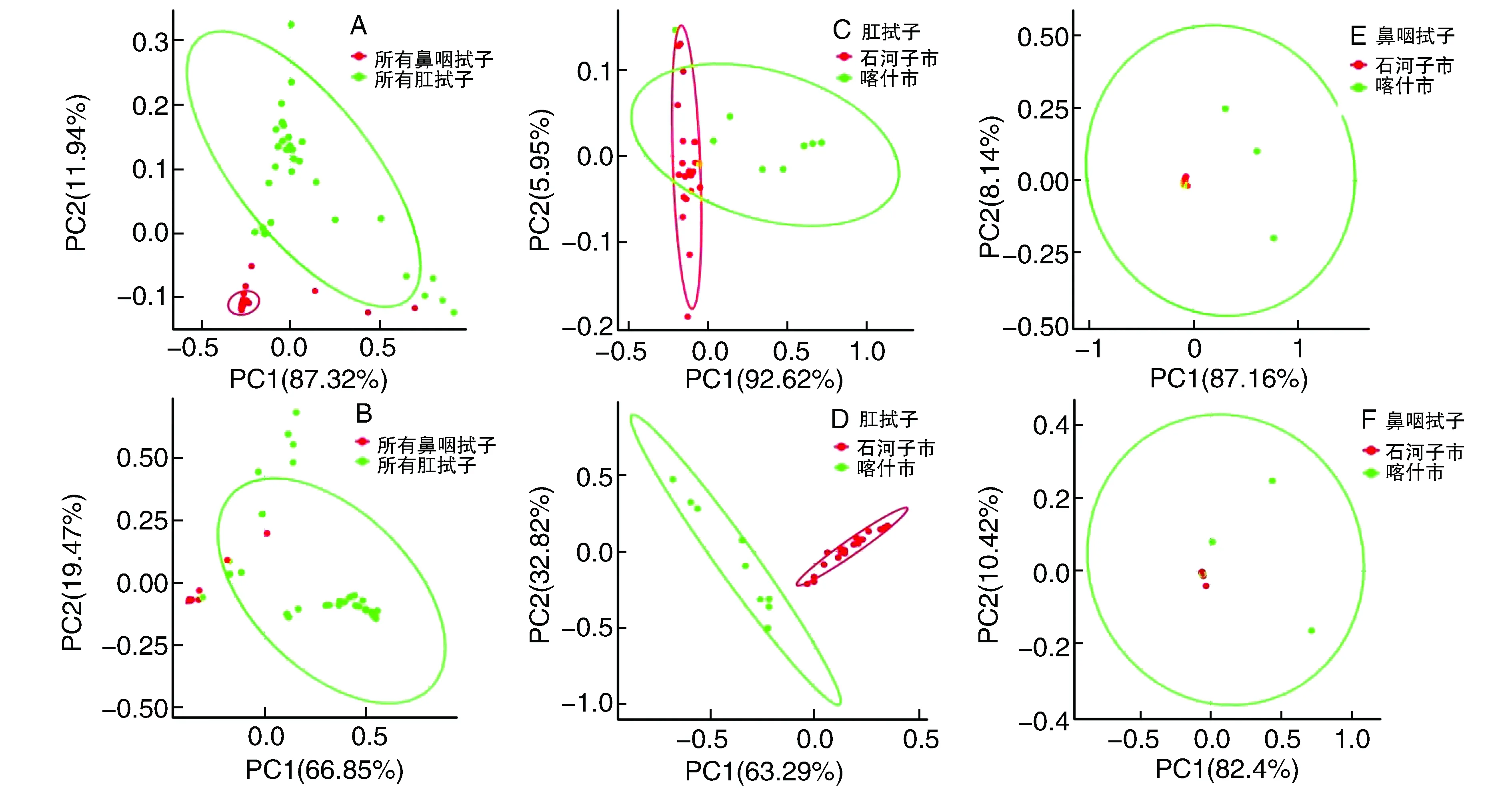
图2 不同类别牛样品细菌物种的PCoA分析A、C和E为门分类水平计算Euclidean距离;B、D和F为属分类水平计算Bray距离。
在微生物分析中,进行完PCoA分析后,采用Anosim分析和Adonis分析不同类别牛样品的细菌种类差异,分别在门水平和属水平两个层级上进行比较分析,差异均具有统计学意义(P<0.05;表1),且组间差异大于组内(R趋向于1;表1)。
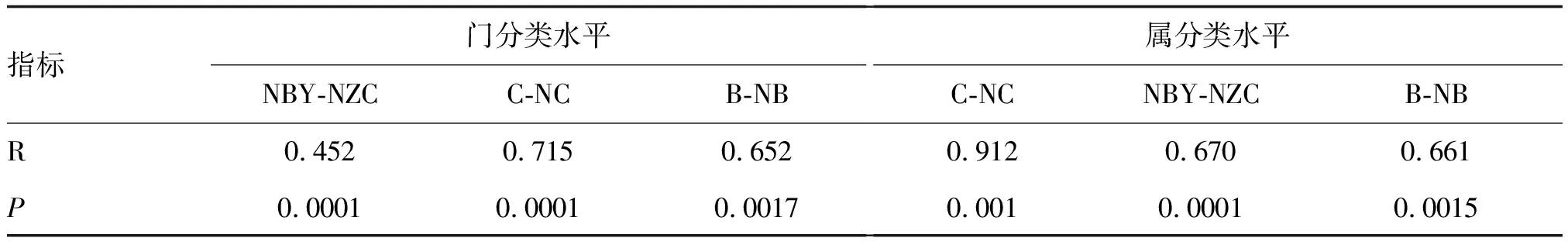
表1 不同类别牛样品的ANOSIM分析和Adonis分析
利用Wilcoxon秩和检验分析所有牛样品的细菌差异种类,在门水平上差异有显著性的菌门共3种,分别是放线菌门、厚壁菌门和拟杆菌门。在属水平上差异有显著性的菌属共16种(图3)。
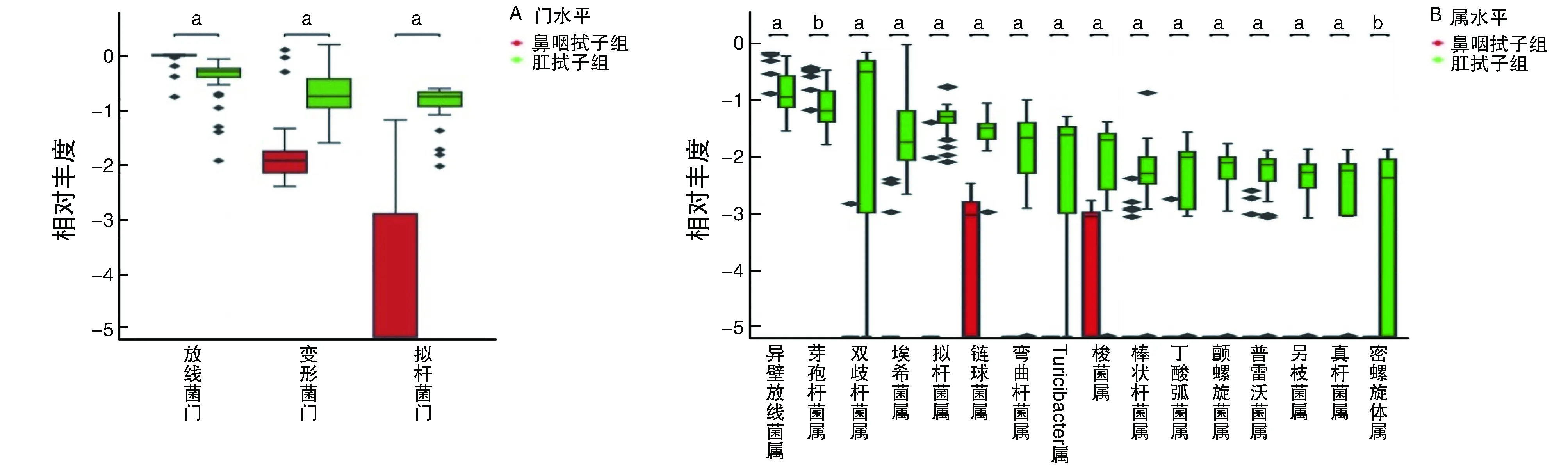
图3 不同牛样品细菌群落差异分析a为P<0.05,b为P<0.01,与鼻咽拭子组比较。
2.4 牛样品中潜在人兽共患病细菌病原菌种类
2.4.1 不同类别牛样品中潜在人兽共患病细菌病原菌种类的比较 牛样品共检出9种人兽共患病原菌(图4),分别是蜡样芽孢杆菌(Bacillus cereus)、大肠杆菌(Escherichia coli)、猪链球菌(Streptococcus suis)、肺炎链球菌(Streptococcus pluranimalium)、鲍曼不动杆菌(Acinetobacter baumannii)、肺炎克雷伯氏菌(Klebsiella pneumoniae)、空肠弯曲杆菌(Campylobacter jejuni)、化脓性链球菌(Trueperella pyogenes)和多杀巴斯德杆菌(Pasteurella multocida)。其中,猪链球菌、空肠弯曲杆菌和化脓性链球菌只在肛拭子样品中检出,鲍曼不动杆菌只在鼻咽拭子中检出。不同类别牛样品中共检出的蜡样芽孢杆菌、大肠杆菌和肺炎链球菌差异存在显著性(P<0.01),而肺炎克雷伯氏菌和多杀巴斯德杆菌差异无统计学意义(P>0.05)。
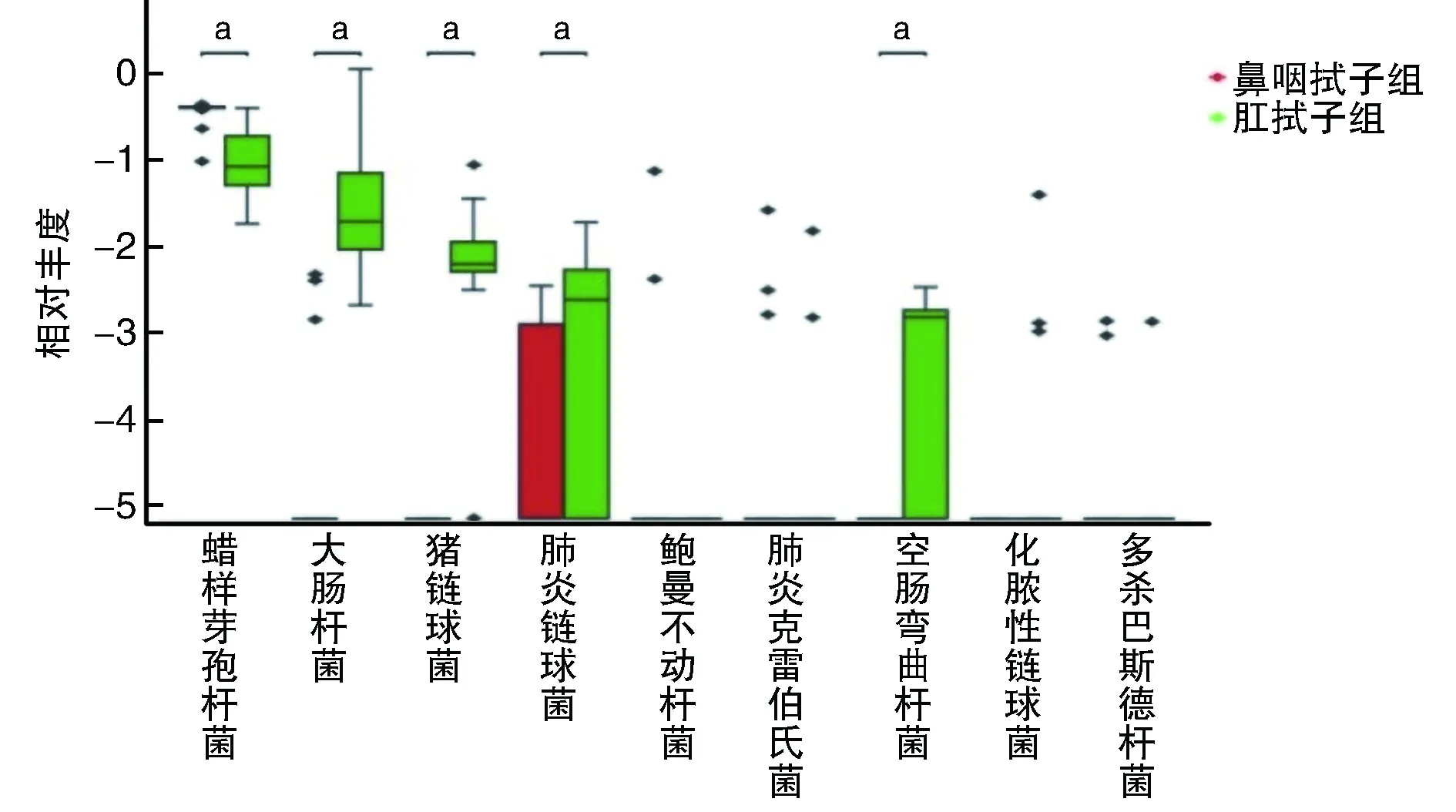
图4 不同类别牛样品潜在人兽共患病细菌病原菌种类的Wilcoxon秩和检验a为P<0.01,与鼻咽拭子组比较。
2.4.2 不同地区牛样品中潜在人兽共患病细菌病原菌种类的比较 Wilcoxon秩检验结果发现,喀什市肛拭子样品中的大肠杆菌和肺炎链球菌相对丰度极显著高于石河子市(P<0.01);喀什市肛拭子样品中的蜡样芽孢杆菌和猪链球菌相对丰度显著高于石河子市(P<0.05)。两个地区鼻咽拭子样品中共检出的蜡样芽孢杆菌和肺炎链球菌差异无统计学意义(P>0.05;图5)。
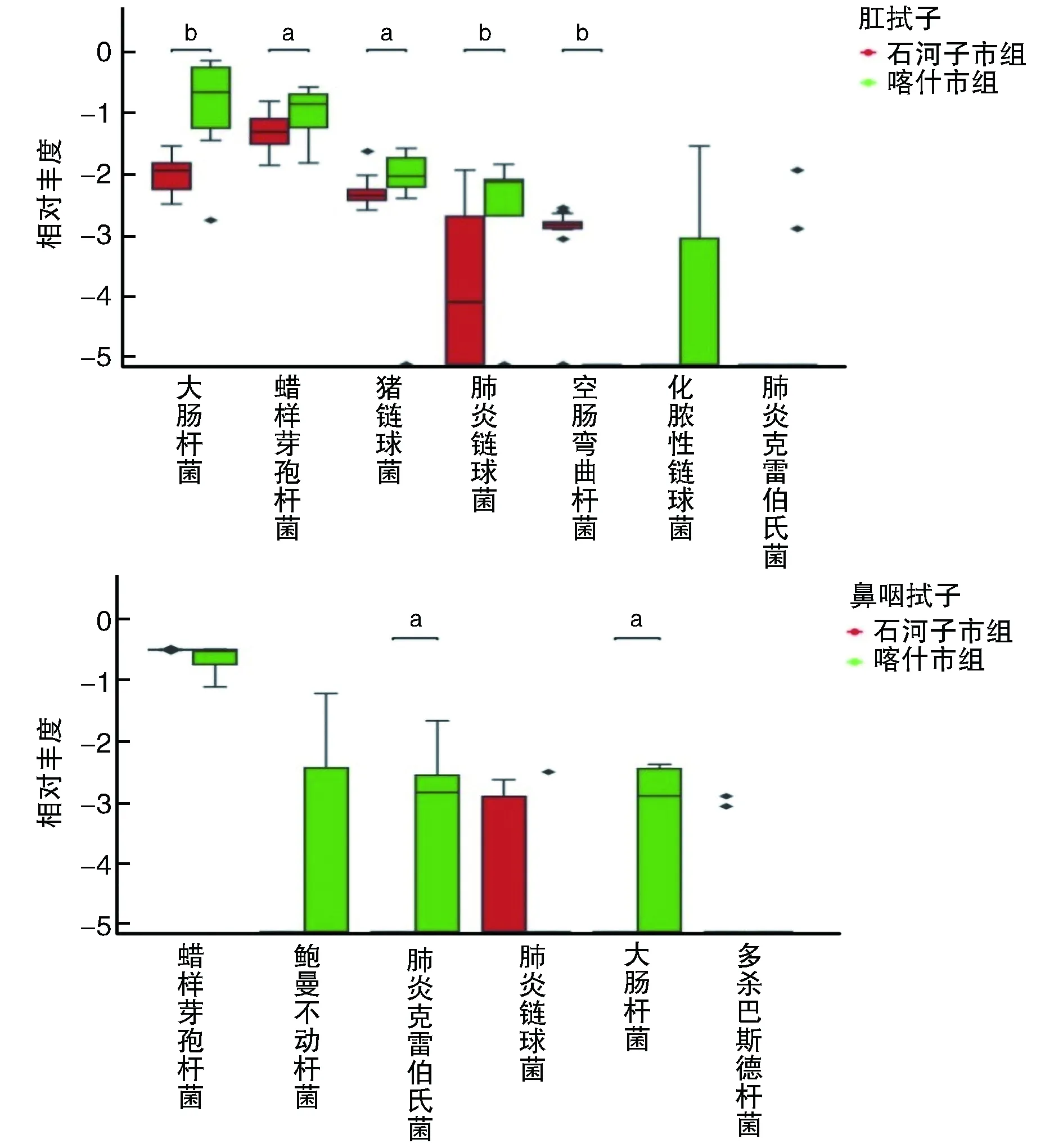
图5 不同地区潜在人兽共患病细菌病原菌种类的Wilcoxon秩和检验a为P<0.05,b为P<0.01,与石河子市组比较。
3 讨 论
目前国内外研究大多都是基于16S高通量测序对牛乳房炎以及牛瘤胃/牛鼻咽微生物区系的报道[11-13],而关于牛携带潜在人兽共患病原菌及细菌菌群多样性全面鉴定分析的研究尚未有报道。不同的检测方法和样本对微生物结构的分析各有不同,但利用先进的二代高通量测序可对样本中全部的微生物多样性、种群结构等进行全面分析,进而可以最大限度地挖掘微生物资源,也可以进行传染病病原溯源和潜在病原快速高通量调查[14]。
本研究应用宏基因组二代测序技术,以牛鼻咽拭子和肛拭子样品提取DNA对新疆南疆和北疆规模化养殖牧场牛潜在病原菌携带状况进行调查分析,通过对牛样品的细菌群落结构多样性分析发现,新疆地区牛肠道和牛呼吸道中微生物主要菌门均以厚壁菌门、放线菌门、变形菌门和拟杆菌门4种为优势菌门。既往国内外研究不同品系的牛瘤胃微生物结果显示主要菌种均是拟杆菌和厚壁菌[12,15],而本研究牛肠道以放线菌门和变形菌门占主导,厚壁菌和拟杆菌次之,这一差异可能是因为牛的生活地域和环境,又或是喂养饲料的不同造成其肠道细菌菌门结构的不同。本研究牛呼吸道中优势菌门与Zeineldin等[13]结果一致,但优势菌属各不相同,这可能是因为养殖环境不同,导致上呼吸道不断暴露于周围环境中的多种细菌中[16]。在物种相对丰度的基础上,本研究也进行了相似性和差异性分析,结果发现牛呼吸道和肠道细菌菌群有一定的相似性,但差异存在显著性(P<0.05),并且组间差异大于组内差异。本研究表明,新疆规模化养殖牧场牛携带的细菌群落相对丰富,且不同样品组间存在差异。
另外,本研究对新疆南疆喀什市和北疆石河子市两个地区的细菌菌系作比较分析,两地区的牛肛拭子样品在门水平上均表现出丰富细菌菌群,但随着分级水平的降低,养殖场的细菌组成差别不断增加,在属水平上两地区各类细菌在数量及分布上有显著差异;两地区鼻咽拭子样品组在门和属水平上均反映出养殖场细菌群落结构的多样性。差异性分析结果表明两个地区的牛肛拭子样品和牛鼻咽拭子样品均是组间差异大于组内(P<0.05)。究其两地区呈现出差异的原因,可能与宿主携带的微生物群落结构与其遗传背景、生长环境、生活饮食和免疫状况等众多因素有关[17]。
本次测序,除检测到大量常规菌外,还从样品中检出了人兽共患病致病菌。本研究在牛样品中鉴定发现有9种人兽共患病原菌,其中大部分都是食源性细菌性致病菌[18-20],虽然目前这些菌种在动物的感染报道较少,但是在某些饲养条件不利因素诱发下,这些条件致病菌可能大量繁殖,导致宿主发病,所以其可能成为人兽共患病传染病的潜在病原,应引起高度重视。还有值得注意的是,不管基于样品类型还是地区水平,蜡样芽孢杆菌和肺炎链球菌均有分布。蜡样芽孢杆菌被认为是一种常见的食品污染物,可以引起人和动物产生呕吐和腹泻等临床症状[21]。肺炎链球菌是一种革兰氏阳性人兽共患病机会致病菌,其是一种新兴的动物链球菌,与牛和禽类原发感染相关[22]。牛源细菌性人兽共患病致病菌不仅影响牛自身的安全,还可能对牧民以及通过牛源食品对大众的健康产生威胁,因此对这类疫病的防控也尤为重要。
综上,本研究对新疆地区牧场牛潜在病原菌携带状况进行调查,总结其细菌群落结构的多样性和牛源人兽共患病致病菌的菌种,并分别比较分析了不同样品和不同地区牛携带菌群的差异性。在后续研究中,可以比对不同的数据库挖掘更多的数据,分析病毒的分布情况;结合牧民的流行病学资料和人兽共患病致病菌的疾病特征作综合分析,评价传播和感染的风险,旨在为牛的规模化养殖和牛源食品的安全等提供更多的科学支持。
