生地黄配方颗粒工艺优化及其量值传递研究*
2021-05-29王雯梁馨月薛佩芸
杨 锦,王雯,梁馨月,薛佩芸,陈 萍,△
(1.陕西中医药大学,陕西 咸阳 712046; 2.兵器工业卫生研究所,陕西 西安 710065; 3.陕西省中医药研究院,陕西 西安 710003)
地黄又名地髓、苄、芑[1],为玄参科多年生草本植物地黄Rehmannia glutinosa Libosch.的根,始载于《神农本草经》,并被列为上品,是传统的四大怀药之一,也是我国传统大宗中药材。生地黄味甘,性寒,归心、肝、肾经,为清热凉血之品,有清热凉血、养阴生津功效,用于热入营血、温毒发斑、吐血等证[2]。其临床主要用于心脑血管疾病[3]、糖尿病及其并发症、骨质疏松等疾病的治疗,并可发挥神经保护作用[4]。生地黄主要含有苯乙醇苷类、环烯醚萜类、糖类、紫罗兰酮类等化合物[5]。目前对其研究主要集中在饮片及其颗粒的质量控制、化学成分、药理学等方面[6],而对生地黄配方颗粒工艺优化及其化学成分的量值传递研究尚未见报道。本研究中采用正交试验法,以出膏率、总多糖及苯乙醇苷类4个成分含量综合值作为评价指标,优选生地黄标准煎剂的最佳提取工艺;饮片经最佳工艺提取,制备成配方颗粒,通过特征成分的含量测定,考察饮片、中间体和配方颗粒间量值传递效果。现报道如下。
1 仪器与试药
1.1 仪器
Agilent1260型高效液相色谱仪(美国Agilent公司);KQ-500DE型数控超声波清洗器(昆山市超声仪器有限公司);CP2102型电子天平(奥豪斯仪器<常州>有限公司,精度为0.01 g);SQP型电子天平(规格为QUINTIX224-1CN、精 度 为0.1 mg,规 格 为SECURA125-1CN、精度为0.01 mg),均购自赛多利斯科学仪器<北京>有限公司;DK-98-Ⅱ型电热恒温水浴锅(天津市泰斯特仪器有限公司);CS101-3EB型电热鼓风干燥箱(重庆四达试验设备有限公司);ZF-C型三用紫外分析仪(上海康禾光电仪器有限公司);Mb型可调式电热板(北京科伟永兴仪器有限公司);ZDHW型调温电热套(北京中兴伟业仪器有限公司,规格为3 000 mL);GZL100-25L型湿法制粒机(石家庄科源机械设备有限公司,功率为7.5 kW);Synergy型超纯水制水机(默瑞<上海>生物科技有限公司)。
1.2 试药
生地黄配方颗粒3批(杨凌生物医药科技股份有限公司,编号分别为01,02,03,批号分别为20200701,20200702,20200703);毛 蕊 花 糖 苷 对 照 品(批 号 为1530-200202,规格为20 mg,含量>98%),D-无水葡萄糖对照品(批号为110833-201908,含量>99.8%),均购自中国食品药品检定研究院;焦地黄苯乙醇苷A1(批号为P16M11S109726,规格为10 mg,含量>95%),焦地黄苯乙醇苷B1(批号为P16M11S109727,规格为20 mg,含 量>97%),异 毛 蕊 花 糖 苷(批 号 为W17J10C90785,规格为20 mg,含量>98%),洋地黄叶苷C(批号为P14M11F109729,规格为5 mg,含量>95%),均购于上海源叶生物科技有限公司;乙腈、磷酸均为色谱纯,甲醇为分析纯,水为自制超纯水;生地黄饮片3批(陕西兴盛德中药饮片有限公司,批号分别为20190101,20190301,20200101),经陕西省中医药研究院中药研究所炮制鉴定室杨智峰研究员显微鉴别均为正品。
2 方法与结果
2.1 指标成分考察
2.1.1 干膏率
分别精密量取煎煮液20 mL,分别置已干燥至恒重的蒸发皿中,水浴蒸干,残渣于105℃干燥3 h,置干燥器中冷却30 min,迅速精密称定质量,计算干膏率。
2.1.2 总多糖含量
溶液制备:称取105℃下干燥至恒重的D-无水葡萄糖对照品适量,精密称定,加水制成质量浓度为1.737 2 mg/mL的葡萄糖对照品溶液。精密量取煎煮液各2.0 mL,分别加入8.0 mL无水乙醇,涡旋混匀,放置过夜,离心,倾出上清液,沉淀,用10 mL 80%乙醇洗涤2次,用水溶解,并定容至10mL,即得供试品溶液。
线性关系考察:分别精密量取葡萄糖对照品溶液0,0.2,0.4,0.6,0.8,1.2,1.6,2.0,3.0 mL,置50 mL容量瓶中,加水定容,摇匀,制成系列对照品溶液,各精密量取3.0 mL,分别置10 mL容量瓶中,加入5%苯酚溶液1.0 mL,涡旋混匀,迅速滴加浓硫酸6.0 mL,涡旋混匀,于50℃水浴放置40 min,取出,置水中冷却至室温,混匀,制成质量浓度分别为0,2.084 6,4.169 3,6.253 9,8.338 5,12.507 8,16.677 1,20.846 4,31.269 6μg/mL的系列待测溶液,按分光光度法(2020年版《中国药典(一部)》附录ⅤA),于490 nm波长处测定吸光度,以葡萄糖质量浓度(X,μg/mL)为横坐标、吸光度(Y)为纵坐标进行线性回归,得回归方程Y=0.045 8 X-0.021 2,R2=0.998 8(n=9)。结果表明,葡萄糖质量浓度在2.084 6~31.269 6μg/mL范围内与吸光度线性关系良好。
吸光度测定:精密量取上述供试品溶液各1.0 mL,加水2.0 mL,同线性关系考察项下方法自“加入5%苯酚溶液1.0 mL”起操作,依法测定吸光度,并计算。
2.1.3 苯乙醇苷类成分含量
色谱条件:色谱柱为Diamonsil Plus C18柱(250 mm×4.6 mm,5μm);流动相为乙腈(A)-0.1%磷酸溶液(B),梯度洗脱(0~10 min时10%A→14%A,10~32 min时14%A→20%A,32~35 min时20%A→10%A,35~37 min时10%A);流速为1.0 mL/min;检测波长为334 nm;柱温为30℃;进样量为10μL。
溶液制备:分别取焦地黄苯乙醇苷A1、毛蕊花糖苷、焦地黄苯乙醇苷B1和异毛蕊花糖苷对照品适量,精密称定,加乙腈-0.1%磷酸溶液(16∶84,V/V),制成每1 mL分别含焦地黄苯乙醇苷A1、毛蕊花糖苷、焦地黄苯乙醇苷B1和异毛蕊花糖苷22.80,34.82,59.36,45.36μg的混合对照品溶液。精密吸取煎煮液各5 mL,置25 mL容量瓶中,分别加甲醇15 mL,超声10 min,用甲醇定容,摇匀,滤过,精密量取续滤液10 mL,蒸干,残渣用乙腈-0.1%磷酸溶液(16∶84,V/V)溶解,移至5 mL容量瓶中,用乙腈-0.1%磷酸溶液(16∶84,V/V)定容,摇匀,滤过,取续滤液,即得供试品溶液。精密吸取5 mL水,按供试品溶液制备方法制备阴性对照品溶液。
系统适用性试验:分别精密吸取混合对照品溶液、供试品溶液、阴性对照品溶液各10μL,按拟订色谱条件进样测定。结果焦地黄苯乙醇苷A1、毛蕊花糖苷、焦地黄苯乙醇苷B1和异毛蕊花糖苷的保留时间分别为20.935,27.399,29.425,31.183 min,理论板数均大于5 000,分离度均大于1.5,拖尾因子为1.16~1.23,详见图1。
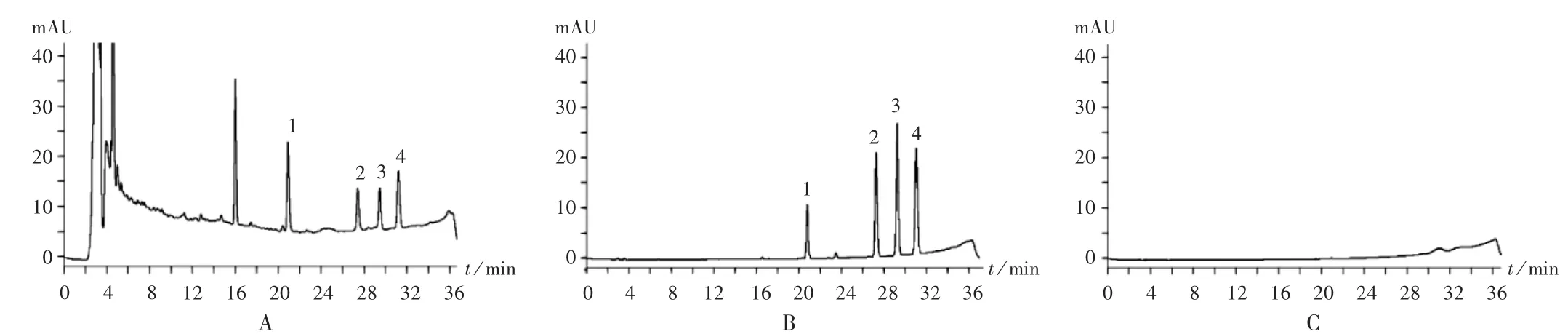
图1 高效液相色谱图Fig.1 HPLC chromatograms
线性关系与检测限、定量限考察:分别精密量取混合对照品溶液0.2,0.5,0.8,1.0,1.5,2.0 mL,置2 mL容量瓶中,用乙腈-0.1%磷酸溶液(16∶84,V/V)定容,摇匀,分别精密吸取10μL,按2.1.3项下色谱条件进样测定,以各成分质量浓度(X,μg/mL)为横坐标、相应峰面积(Y)为纵坐标进行线性回归,得回归方程、相关系数和线性范围,以信噪比(S/N)为3和10时的质量浓度分别作为检测限和定量限,结果见表1。
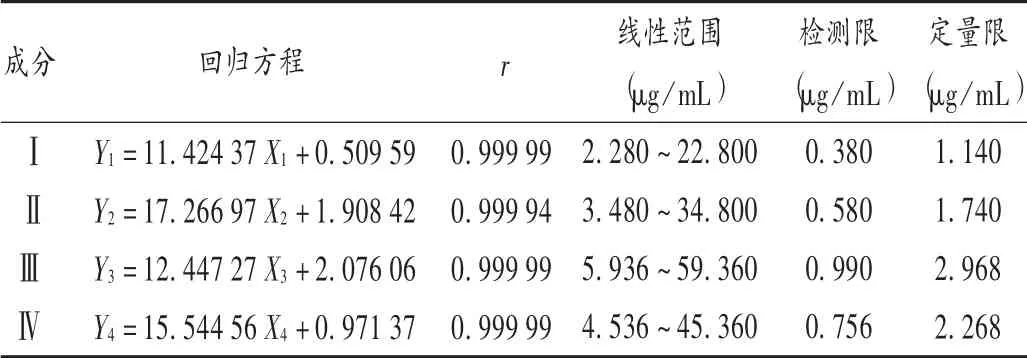
表1 线性关系与检测限、定量限考察结果(n=6)Tab.1 Linear relationship and the results of LOD and LOQ(n=6)
精密度试验:分别精密吸取对照品溶液10μL,按2.1.3项下色谱条件重复进样6次,并连续测定6 d。结果焦地黄苯乙醇苷A1、毛蕊花糖苷、焦地黄苯乙醇苷B1、异毛蕊花糖苷日内、日间峰面积的RSD分别为0.45%,0.45%,0.25%,0.13%(n=6)和0.29%,0.33%,0.27%,0.45%(n=6),表明仪器精密度良好。
稳定性试验:精密吸取供试品溶液10μL,室温下按2.1.3项下色谱条件分别于0,2,4,8,12,16,24 h时进样测定。结果焦地黄苯乙醇苷A1、毛蕊花糖苷、焦地黄苯乙醇苷B1和异毛蕊花糖苷峰面积的RSD分别为1.29%,2.37%,3.66%,2.37%(n=7),表明供试品溶液在室温下放置24 h内稳定。
重复性试验:取煎煮液5 mL,共6份,依法制备供试品溶液,按2.1.3项下色谱条件进样测定。结果焦地黄苯乙醇苷A1、毛蕊花糖苷、焦地黄苯乙醇苷B1和异毛蕊花糖苷的平均含量分别为129.08,283.68,67.82,78.27μg/mL,RSD分别为4.46%,4.03%,4.84%,4.17%(n=6),表明方法重复性良好。
加样回收试验:精密量取已知含量的煎煮液2.0 mL,共9份,分为3组,置25 mL容量瓶中,分别精密加入焦地黄苯乙醇苷A1对照品溶液(237.5μg/mL)0.5,1.0,1.6 mL,毛蕊花糖苷对照品溶液(489.6μg/mL)0.6,1.2,1.8 mL,焦地黄苯乙醇苷B1对照品溶液(148.4μg/mL)0.4,0.9,1.4 mL,异毛蕊花糖苷对照品溶液(113.4μg/mL)0.7,1.4,2.0 mL,加甲醇15 mL,超声10min,用甲醇定容,摇匀,滤过,精密量取续滤液10mL,蒸干,残渣用乙腈-0.1%磷酸溶液(16∶84,V/V)溶解,转移至5 mL容量瓶中,用乙腈-0.1%磷酸溶液(16∶84,V/V)定容,摇匀,滤过,取续滤液,按2.1.3项下色谱条件进样测定,并计算回收率。结果见表2。
2.2 吸水率考察
称取药材样品(批号为20200101)50.0 g,共3份,依次加水600 mL,搅匀,浸泡,分别于20,40,60,80,100 min及2,12,13 h时滤出药液,读取体积,直至体积不再减少。结果表明,药液体积12 h后不再增加,计算吸水率为185.9%,故首次加水量不得低于10倍量。
2.3 正交试验与结果[7-9]
因素水平:影响样品水煎煮的主要因素有浸泡时间、煎煮时间、加水量(倍数)、煎煮次数及粒度,考虑大生产常以饮片投料,在预试验基础上,选择浸泡时间(A)、煎煮时间(B)、加水量(C)、煎煮次数(D)作为考察因素,以干膏率、总多糖及苯乙醇苷类4个成分含量综合值为考察指标,因素水平见表3。
正交试验及方差分析:结果见表4、表5。可见,各因素作用强弱依次为D>A>C>B;因素A和因素D对结果有显著影响。根据大生产实际情况综合考虑,确定最佳提取工艺为A3B2C2D3,即浸泡1.5 h,分别加10,8,6倍量水,3次分别煎煮2.0,1.5,1.0 h。

表2 加样回收试验结果(n=9)Tab.2 Results of the recovery tests(n=9)

表3 因素水平表Tab.3 The factors and levels

表4 正交试验设计与结果Tab.4 Design and results of the orthogonal test
验证试验:取药材样品(批号为20200101)50.0 g,共3份,按最佳工艺进行煎煮、浓缩,稀释至1 000 mL,制备成供试品溶液。结果供试品溶液中生地黄总多糖、焦地黄苯乙醇苷A1、毛蕊花糖苷、焦地黄苯乙醇苷B1、异毛蕊花糖苷含量及干膏率均大于或接近正交试验表中的最高值,且重复性好,工艺相对稳定、合理。详见表6。

表5 方差分析结果Tab.5 Results of ANOVA

表6 验证试验结果(n=3)Tab.6 Results of the verification test(n=3)
2.4 成型工艺研究
采用减压浓缩方法,生地黄提取物以60℃减压(-0.09 MPa)条件浓缩至相对密度为1.10~1.20(60℃)的溶液,以糊精作为辅料,以浸膏与糊精(2.5∶1,m/m)混合,乙醇制粒,干燥,即得配方颗粒。
配方颗粒最佳制备工艺:取药材2.0 kg,以水浸泡2 h,分别加10,8,6倍量水,分别煎煮2.0,1.5,1.0 h,滤过,合并滤液,浓缩至相对密度为1.20~1.30(60℃)的稠膏,加适量糊精,浸膏与糊精(2.5∶1,m/m)混合,以乙醇为黏合剂制粒,干燥,即得配方颗粒1.0 kg。
2.5 中试试验[7]
取3批药材样品各20.0 kg,以2.4项下工艺制备,得3批配方颗粒,进行试验。结果见表7。

表7 中试试验结果(n=3)Tab.7 Results of the Pilot test(n=3)
2.6 指标成分量值传递[8-9]
对照品制备:分别精密量取2.1.3项下混合对照品溶液1.0 mL,置2 mL容量瓶中,用乙腈-0.1%磷酸溶液(16∶84,V/V)定容,制成每1 mL分别含焦地黄苯乙醇苷A1、毛蕊花糖苷、焦地黄苯乙醇苷B1和异毛蕊花糖苷11.400,17.408,29.680,22.680μg的混合对照品溶液1。
供试品制备:称取3批药材饮片样品粗粉各约0.8 g,精密称定,置具塞锥形瓶中,精密加入甲醇50 mL,称定质量,加热回流提取1.5 h,放冷,再次称定质量,用甲醇补足减失的质量,摇匀,滤过,精密量取续滤液20 mL,减压回收溶剂近干,残渣用乙腈-0.1%磷酸溶液(16∶84,V/V)溶解,转移至5 mL容量瓶中,用乙腈-0.1%磷酸溶液(16∶84,V/V)定容,摇匀,滤过,取续滤液,即得饮片供试品溶液;分别取中间体、配方颗粒适量,各3批,研细,取约2.0 g,精密称定,同饮片供试品溶液方法制备,即得中间体供试品溶液和配方颗粒供试品溶液。
含量测定:取混合对照品溶液1和上述3种供试品溶液适量,按2.1.3色谱条件进样,测定焦地黄苯乙醇苷A1、毛蕊花糖苷、焦地黄苯乙醇苷B1和异毛蕊花糖苷的含量,计算转移率。结果见表8。
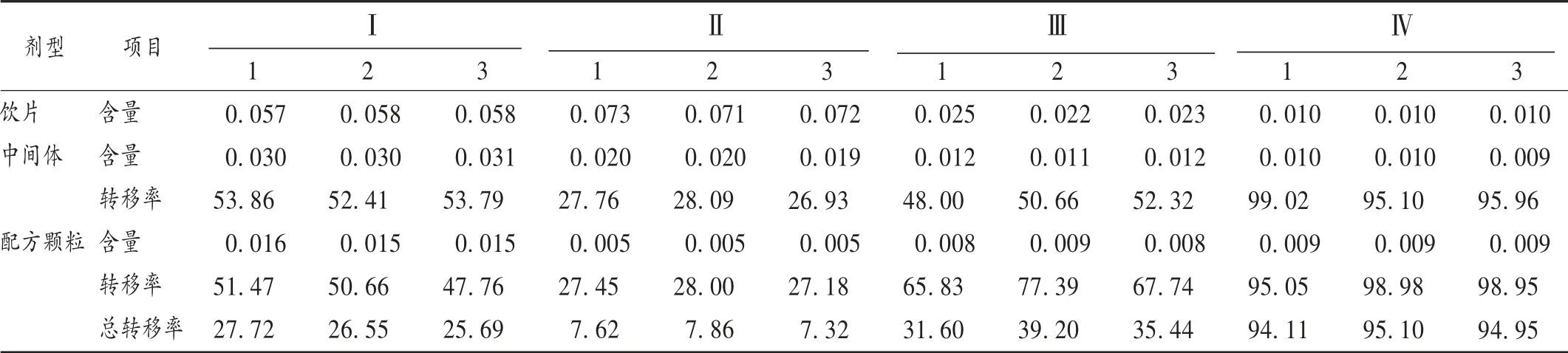
表8 饮片→中间体→配方颗粒的苯乙醇苷类成分量值传递结果(%)Tab.8 Quantity transfer results of phenylethanoid glycosides from decoction pieces→intermediates→formula granules(%)
3 讨论
3.1 流动相选择
预试验中比较了乙腈-0.1%醋酸溶液和乙腈-0.1%磷酸溶液作流动相时对结果的影响,测定苯乙醇苷类含量时,以乙腈-0.1%磷酸溶液为流动相时基线更平稳,特征成分分离效果更好。故优选乙腈-0.1%磷酸溶液作为洗脱体系,并优化得到最佳洗脱程序。
3.2 成分量值转化分析
生地黄饮片、正交煎煮液、中间体、配方颗粒中均检测到洋地黄叶苷C成分,其保留时间为16.035 min,分离度较好,由于对照品量少且不稳定,配制的溶液保存时间短,不易进行含量测定,故只作成分指认;中药在煎煮和制剂工艺过程中可能产生新物质或发生其他动态变化,是中药药效物质基础研究中的重要因素。本试验中,生地黄中间体和配方颗粒中的毛蕊花糖苷转移率偏低,异毛蕊花糖苷转移率偏高,由于毛蕊花糖苷和异毛蕊花糖苷为同分异构体,生地黄在煎煮过程中毛蕊花糖苷可能部分转化为异毛蕊花糖苷[10-12],将焦地黄苯乙醇苷A1、毛蕊花糖苷、焦地黄苯乙醇苷B1和异毛蕊花糖苷4个成分含量综合值作为评价生地黄的量值传递研究的特征成分更合理。
3.3 辅料选择
采用优选的最佳提取工艺进行生地黄特征成分的提取,能提高制剂含药量,利于饮片发挥药效;生地黄饮片中特征成分的化学性质不稳定,采用60℃减压(-0.09 MPa)浓缩至相对密度为1.20~1.30(60℃);由于中药浸膏多具有吸湿性,故选择加入适当、适量的辅料与之混合,辅料选用糊精、淀粉进行考察,其中糊精具有无异味、溶解性好、不易吸潮、稳定性好等优点,可溶性淀粉具有成型性好、化学性质稳定且吸附力强、流动性好等优点[13-14]。本研究结果显示,糊精比淀粉流动性、成型性好,不易吸湿,故选择糊精作为辅料。又考察了润湿剂为乙醇溶液时,浸膏与糊精的比例分别为3∶1(m/m)和2.5∶1(m/m)时的生地黄配方颗粒质量,结果确定浸膏与糊精的比例为2.5∶1(m/m)时生地黄配方颗粒质量最佳。
传统的中药煎剂制备成中药配方颗粒后,由于剂型发生改变,可能影响特征成分的量值传递。在制备工艺过程中,综合考虑苯乙醇苷类4个成分的总体含量、总干膏率均稳定,转移率高,生地黄饮片-中间体-配方颗粒间特征成分的量值转化过程较好地保留了原饮片成分,一致性较好,本研究为配方颗粒替代传统煎剂的研究提供了思路。
