尘埃落定
——1例进行性眼外肌麻痹患者的诊疗
2021-05-27孙平杨仕林朱雯华卢家红陈倩田国红
孙平 杨仕林 朱雯华 卢家红 陈倩 田国红
(1复旦大学附属眼耳鼻喉科医院眼科 上海 200031;2复旦大学附属华山医院神经内科上海 200040)
慢性进行性眼外肌麻痹(chronic progressive external ophthalmoplegia, CPEO)为一组以慢性进展性双侧上睑下垂、眼球运动广泛障碍为主要临床特征的线粒体疾病。由于导致眼肌麻痹的疾病种类繁多,很多神经系统肌源性病变及神经肌肉接头病变均可累及眼外肌,表现为相似的眼部症状。本文通过对1例以上睑下垂首诊眼科的CPEO患者的临床特征分析,希望加强眼科医师对该类疾病的早期识别和正确处理。
1 病例
患者女性,43岁,因进行性双侧眼睑下垂数年来诊。患者于7~8年前发现双眼上睑下垂,并呈现缓慢进展趋势。无明显视物成双及晨轻暮重的现象。否认家族中类似病史及其他遗传病史。神经眼科查体:神清、语利。最佳矫正视力:右眼0.7,左眼0.9。双侧睑裂高度5 mm。双眼上睑下垂,遮盖瞳孔2/3。双眼水平与垂直运动均受限(图1)。前节未见异常,双侧瞳孔等大等圆,对光反射灵敏,眼底双侧视盘边界清,右侧视盘色略淡,未见视网膜色素样改变(图2)。神经系统查体:双侧咀嚼肌力略减弱。左侧鼻唇沟变浅。双眼闭目正常。吞咽无困难。四肢除下肢屈髋肌力4+级,余肢体肌力5级,深浅感觉、腱反射均正常,病理征未引出。眼眶增强磁共振成像(magnetic resonance imaging,MRⅠ)示双侧眼外肌弥漫变细,眶内脂肪填充(图3)。线粒体全基因(mitochondrial DNA,mtDNA)检查未见异常,与线粒体功能相关的核基因MYH2基因突变(c.2266G>A),提示包涵体肌病。将患者转诊至神经科进一步检查。肌肉MRⅠ示双侧臀大肌、股二头肌长头、半腱肌肌萎缩、脂肪浸润。肌电图示轻度肌源性损害肌电改变。血糖及血乳酸在正常范围。左侧肱二头肌肌肉活检,免疫组织化学染色发现破碎红纤维(ragged-red fiber)、琥珀酸脱氢酶(succinate dehydrogenase,SDH)过度表达(破碎蓝纤维,ragged-blue fiber)及细胞色素氧化酶(cytochrome oxidase,COX)染色阴性(图4)。肌肉组织送检基因检测发现线粒体mtDNA全长m12127-14377单重缺失,符合线粒体基因组的单重缺失综合征。最终诊断为CPEO。患者后于整形科行上睑下垂手术,采用额肌悬吊术式,术后外观满意(图5),远期效果继续随访中。
2 讨论
CPEO是以双侧眼睑下垂、眼球运动弥漫受限,或合并其他系统性疾病的线粒体功能障碍所导致的一组疾病。CPEO合并其他系统病变时称为CPEO叠加综合征(CPEO plus syndrome)。早在1868年,von Grafe 即 报 道 了1例CPEO病例,但发病机制及基因学改变不详。数十年后通过对CPEO患者眼外肌及四肢肌肉的组织化学分析发现了破碎红纤维(肌纤维内线粒体的异常聚集),提示该类疾病为线粒体疾病。随着基因检测技术的发展,mtDNA大片段缺失、点突变及与线粒体功能密切相关的核基因突变进一步证实CPEO及CPEO叠加综合征是一类线粒体疾病[1]。

图1 慢性进行性眼外肌麻痹患者九眼位图 双侧眼睑下垂、眼球水平运动及垂直运动均明显受限。
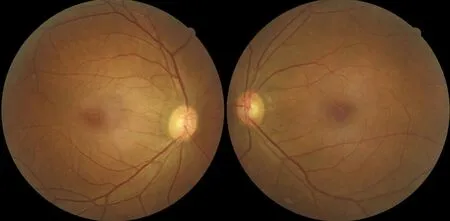
图2 双眼底视盘 边界清,右眼视盘色略淡,左眼视盘色红,未见视网膜色素样改变。

图3 眼眶MRI 眼外肌弥漫萎缩变细:A.轴位T1WⅠ增强扫描;B.冠状位T1WⅠ增强扫描;C.冠状位T2WⅠ。
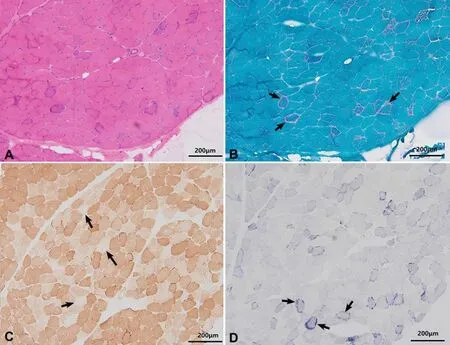
图4 左侧肱二头肌肌肉活检 A. HE染色;B. Gomori 三色染色示破碎红纤维(箭头);C. COX染色示COX阴性纤维(箭头);D. SDH 染色示过度反应的破碎蓝纤维(箭头)。
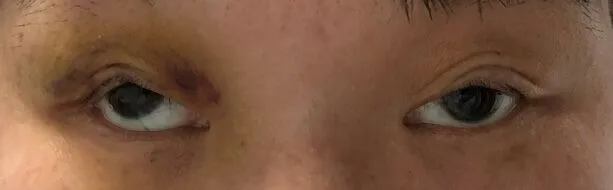
图5 患者行额肌悬吊上睑下垂手术后外观改善
CPEO眼部症状可发生于任何年龄,通常起病隐匿,并呈现进行性加重。双眼对称性上睑下垂是CPEO患者最易发觉的症状,部分患者可因上睑下垂而采取抬头仰颌的代偿头位。临床偶见上睑下垂呈现非对称性。疾病早期,眼外肌麻痹表现为各方向扫视运动迟缓、不到位。随病情恶化,晚期眼肌麻痹呈完全性。由于眼外肌对称性广泛受累,且病变慢性进展产生单眼抑制,因此患者少有复视的主诉,或复视并不是就诊的突出症状。斜视检查通常能够发现眼球各向运动不充分,尤其是水平运动较垂直运动障碍更加明显,且外斜视较内斜视更为常见。幼年发病患者严重的上睑下垂及斜视可导致弱视。患者瞳孔不受累。患者也可出现眼轮匝肌、面肌及四肢肌肉无力[2]。
CPEO基因突变具有相当大的异质性。50%为散发型,以mtDNA单重大片缺失为代表,且并不遗传给下一代。同样的大片段缺失还可导致临床表现各异的线粒体病变,如Kearns-Sayre综 合 征(Kearns-Sayre syndrome,KSS)。作 为CPEO的一个特殊类型,KSS患者除眼外肌麻痹外,尚伴有色素性视网膜病变及心脏传导阻滞,于1958年被Kearns和Sayre报道[3]。近期研究[4-5]显示,不同的缺失位点与缺失范围与CPEO及多种CPEO叠加综合征的临床表型密切相关。另外50%的CPEO患者可为常染色体显性(autosomal dominant,AD)、常染色体隐性(autosomal recessive,AR) 或母系遗传的方式。其中核基因POLG1、POLG2、ANT1及OPA1等为常见基因突变。这些基因产物对线粒体稳定性维持及复制起重要作用,突变导致线粒体核苷酸代谢异常,进而继发性导致mtDNA的缺失。这些基因突变多见于CPEO或CPEO叠加综合征。表现为痴呆、帕金森、脊髓小脑共济失调、肌阵挛、构音障碍及延髓麻痹(球麻痹)等。
四肢肌肉活检组织常规免疫组织化学染色诊断CPEO的阳性率约75%,其余仍需要进行DNA分析。由于mtDNA片段缺失或点突变,线粒体大量增生聚集,以Gomori trichrome染色肌膜下线粒体堆积红染(破碎红纤维)、SDH过度表达(破碎蓝纤维)及对应的COX染色阴性为主要表现,且后两者对诊断的敏感度更高[6]。在一些罕见AR遗传的CPEO患者中,线粒体存在耗竭而非过度增殖,少见破碎红纤维,但COX染色仍为阴性。肌肉组织同时进行mtDNA基因检测是最佳选择,这是因为骨骼肌组织内突变线粒体所占的比例远高于血细胞及其他组织。
本例患者虽然慢性进行性上睑下垂及眼球运动障碍的临床表现符合CPEO,但血液样本中并未检测到全线粒体基因异常,仅发现与包涵体肌病相关的核基因MYH2突变。但肌肉活检免疫组织化学强烈提示线粒体肌病,且后续肌肉组织送检基因检测符合mtDNA的单重缺失,符合CPEO的最终诊断。因此,针对性的肌肉活检结合基因检测能够大大提高线粒体肌病的确诊率。
该病的鉴别诊断需要考虑其他类型的肌源性病变,包括肌营养不良、眼咽型肌营养不良、眼咽远端型肌营养不良及重症肌无力等。尤其是眼肌型重症肌无力可模拟任何类型的眼肌麻痹,但病程较短,复视主诉突出,且症状具有波动性和疲劳性。先天性眼外肌纤维化虽然属于眼球运动中枢核团发育及支配异常性疾病,但临床常以上睑下垂及眼外肌麻痹首诊眼科,需要高度重视。
CPEO的治疗尚无针对性药物,对于引起视轴遮挡的肌源性上睑下垂,手术是主要的治疗手段。额肌悬吊术是目前主流术式,可以部分程度减轻因Bells现象不足导致的角膜暴露风险。由于CPEO患者调节及融合能力差,随着病情的进展将出现会聚不足及复视的主诉。临床需根据病情配戴三棱镜或采取个体化的斜视矫正方案。
