基于加权基因共表达网络分析食管鳞状细胞癌放化疗的敏感性基因※
2021-05-26郝彦惠冯瑞兴
郝彦惠,冯瑞兴,殷 麟,陈 凡&
(1.青海大学医学院,青海 西宁 810000;2.青海大学附属医院 青海大学附属肿瘤医院,青海 西宁 810000)
对食管鳞状细胞癌(esophageal squamous cell carcinoma,ESCC)采取新辅助放化疗(neoadjuvant chemoradiotherapy,nCRT)结合手术模式被越来越多地应用于临床。ESCC新辅助放化疗的结果具有明显异质性。对nCRT病理学完全反应(Pathological Complete Response,pCR)的患者比无病理学反应(Non Pathological Complete Response,npCR)患者往往表现出更好的预后效果。此外,接受术前放化疗的无反应患者,会遭遇两个临床问题:其一,放射治疗的相关并发症可能影响患者生存质量;其二,由于nCRT延迟手术切除时间可导致肿瘤播散。因此,迫切需要通过寻找基因生物标志物来预测ESCC患者对nCRT的反应来指导临床治疗。
加权基因共表达网络分析(weighted gene coexpression network analysis,WGCNA)是一种定量测量基因间相互连接程度及基因在网络中重要性的系统生物学方法,其通过模块分析发现疾病相关基因、或未知基因。WGCNA利用拓扑重叠差异度进行基因模块预测,使得模块区分的精密性大大提高。同时,用拓扑重叠来描述两个基因的关系具有更好的稳定性,降低了假阳性或假阴性率。因此,WGCNA是检测维持表达模式相似基因的基因模块的有用工具,也是识别疾病生物标志物和基因功能的有用工具[1]。此外,由于使用WGCNA法的基因间假阳性相对较少,因此广泛用于研究复杂疾病,包括子宫内膜癌[2]、精神分裂症[3]和乳腺癌[4]等。
本研究从系统水平的视角,利用pCR组和npCR组的芯片表达谱数据筛选差异表达基因。在此基础上依赖WGCNA手段分别构建两组加权基因共表达网络,通过对比两组共表达网络的差异,并利用基因保守性统计分析方法确定特异性放化疗敏感性基因模块。再结合GO分类分析和KEGG通路富集分析来阐明各个基因模块的生物学作用,为后续研究筛选生物标志物提供依据。
1.资料与方法
1.1 数据预处理与差异基因筛选
于GEO数据库获得术前接受放化疗ESCC的全基因组表达数据(GSE45670),平台为GPL570。其中包括了11个pCR样本和17个npCR样本,患者依次接受了nCRT和手术。放疗具体方案:术前放疗剂量45~50.4 Gy,分25或28次进行。化疗具体方案:经静脉注射长春瑞滨(25mg/m2,于第1、8、22、29d),顺铂(75mg/m2,于第1、22d),或经静脉注射顺铂(25mg/m2)1~4 d和22~25 d。在nCRT结束后大约4~6 w内行手术治疗。
本研究中基因组数据处理使用R 4.0.3(https://www.r-project.org/)软件。使用Affy函数包对CEL文件的多微阵列原始数据进行清洗、校正、归一化,以及数据转换[5-6]。利用WGCNA函数包结合FDR校准准则(P≤0.1)ttest函数包确定pCR组与npCR组差异表达基因(P≤0.05),共获得1 489个差异基因,基于差异基因行基因共表达网络分析[7]。
1.2 加权基因共表达网络的构建和分析
基于差异表达基因,利用R软件WGCNA包中的blockwiseModules函数构建加权基因共表达网络[8]。基于生物分子网络均为无标度网络的事实,加权邻接函数构建关键参数β(被确定为9)[9]。根据pCR和npCR组的加权邻接矩阵的定义,建立拓扑差异度矩阵(TOM)。TOM根据两个基因在整个网络中共享邻接的程度反映它们之间的相对互联性[9]。经过邻接矩阵与TOM的计算、构建,并利用层次聚类分析法获得pCR组和npCR组的基因网络模块,计算各模块的模块特征基因(module eigengene,ME)与疾病性状的关联性确定加权基因共表达网络模块(P≤0.05)。
1.3 模块保守性评估及临床特征关联性分析
本研究将Zsummary和medianRank两种测量方法相结合评估模块保守性。保守性差则表明与ESCC的放化疗敏感性关系密切。利用基因显著性(Gene Significance,GS)和模块显著性(Module Significance,MS)评估基因表达网络模块与pCR组、npCR临床特征间的关系。一般基因网络模块中GS和MS的绝对值越高,表示该基因与放化疗敏感性越强。
1.4 枢纽基因的寻找及分析
枢纽基因为模块中与疾病关系最为密切的基因。模块隶属度(module membership,MM)用来评估基因与特定模块中ME之间的相关性,用以衡量目的模块内的某一基因在该模块中的从属程度及重要性。因此,理想的hub基因在给定的模块内具有最高连接度和最高的MM、最高的GS。
1.5 统计学处理
使用R 4.0.3软件分析。基因网络可视化应用Cytoscape软件实现。
2.结果
2.1 pCR和npCR组加权基因共表达网络的构建
结合t检验(P≤0.05)、FDR校正准则,确定了pCR组与npCR组间的差异基因共1 489个,基于此进行网络构建。利用R软件的WGCNA函数包构建各组加权基因共表达网络。在此基础上,采用动态剪切树算法分割模块,设置模块基因数(最少基因数为30),并合并相似模块。依此在npCR组中检测到8个模块(图1A)、pCR组中检测到5个模块(图1B)。未聚类到任何模块的基因保留在WGCNA函数包中的灰色模块中。
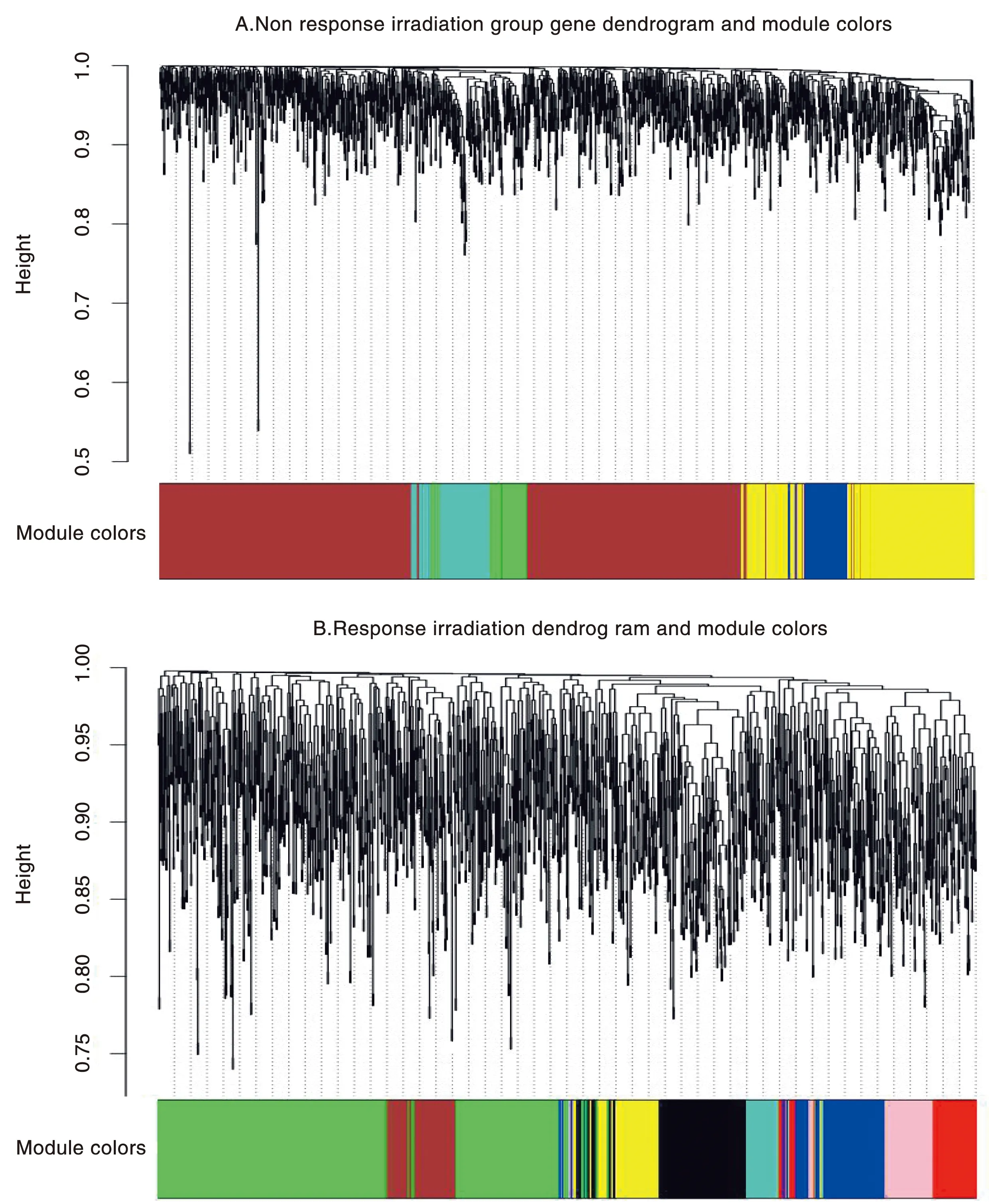
图1 npCR组与pCR组基因共表达模块网络图
2.2 放化疗敏感性相关模块的筛选
结合Zsummary和medianRank两种测量方法对模块行保守性评估,共鉴定出2个Zsummary(≤10且medianRank值较大),分别是yellow模块和blue模块(图2)。另在pCR组探测到black、green、red等三个模块,将上述5个模块作为可能在放化疗敏感性中起重要作用的基因模块。其余brown、turquoise模块保守性好,排除在后续分析之外。

图2 npCR组与pCR组模块保守性分析图
2.3 枢纽基因的确定
本研究中,5个相关模块中MM值与模块内连接度之间呈相关性,说明越靠近网络中心的节点连接度也越高。而GS与连接度并无相关性。因此,本研究以GS、MM值作为筛选枢纽基因的标准,共筛选到5个枢纽基因(图3),分别是MICAL2、EPHB6、COMMD8、EPS15L1、RBPMS。
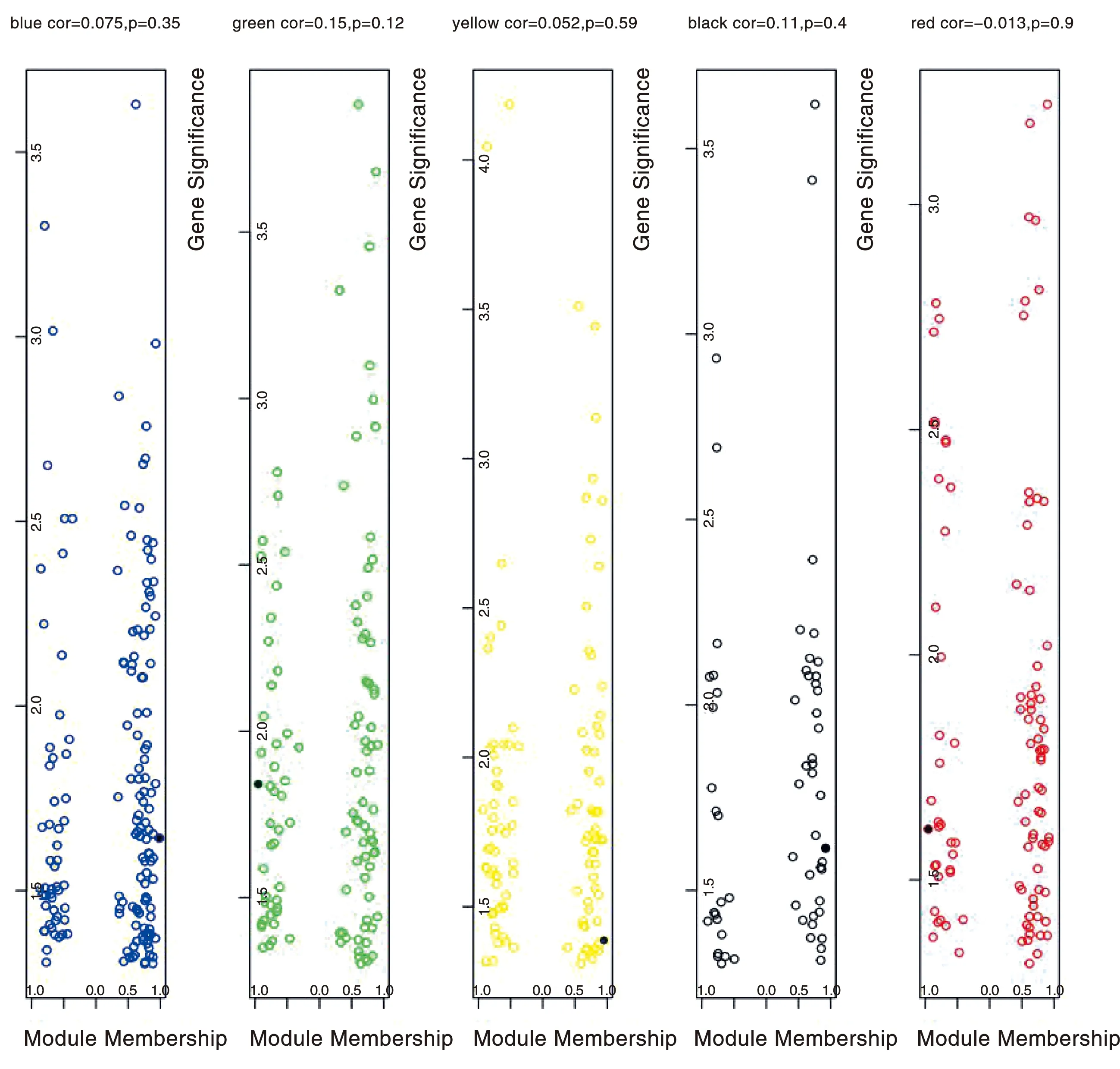
图3 MM与GS的相关性分析图
2.4 基因共表达网络模块的GO、KEGG富集分析
为进一步探索模块的可能功能,对上述5个与放化疗敏感性相关模块的基因进行富集分析。GO富集分析显示(表1),Black模块参与了调节吞噬(GO:0050764)和清除凋亡细胞(GO:0043277)及凝固血液(GO:0007596)等生物学过程。Blue模块参与了内质网的形成(GO:0009954)、细胞生长的正向调控(GO:0030307)、细胞间的连接(GO:005911)、肌细胞的分化(GO:0042692)等生物学过程。Green模块参与了有丝分裂微管细胞骨架的组成(GO:1902850)、有丝分裂纺锤体的组成(GO:0007052)、染色体的分离(GO:0007059)等生物过程。Red模块参与胶原蛋白生物合成和代谢过程的调控(GO:0032965;GO:0010712)、细胞与基质粘附的调控(GO:0010812)等生物过程。Yellow模块参与了内皮细胞凋亡的调控(GO:0072577)、脂质代谢的调控(GO:0006629)等生物过程(表1)。

表1 关键基因网络模块GO富集结果
KEGG通路富集分析显示,Black模块在脂肪酸降解(hsa00071)、氨基酸生物合成(hsa01223)等信号通路显著富集。Blue模块在氧化磷酸化(hsa00190)、唾液分泌(hsa04970)等通路显著富集。Green模块在cAMP信号通路(hsa04024)、p53信号通路(hsa04115)等通路显著富集。Red模块在黑色素瘤(hsa05218)、膀胱癌(hsa05219)等通路显著富集。Yellow模块在cAMP(hsa04024)、cGMP-PKG信号通路(GO:04022)显著富集(表2)。
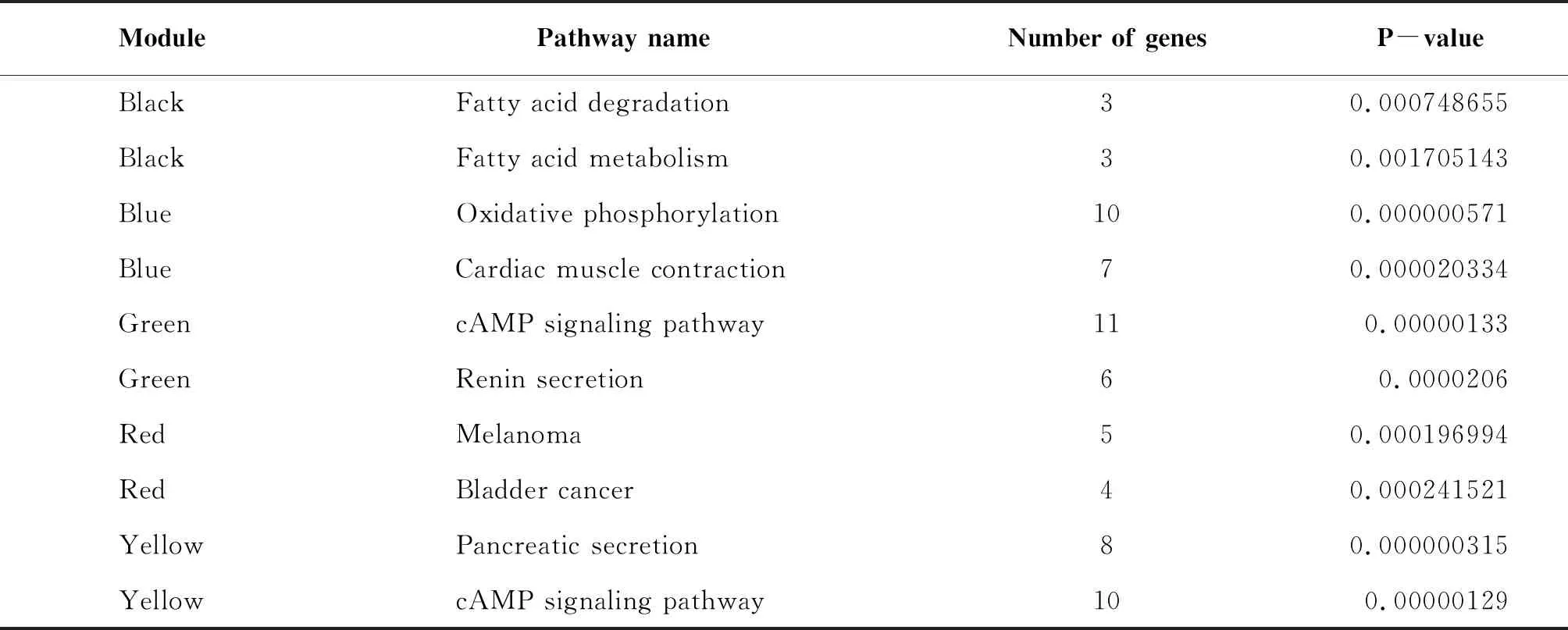
表2 关键基因网络模块KEGG通路富集结果

图4 模块中共表达基因的相互作用图
3.讨论
本研究中共筛选出与食管鳞癌放化疗敏感性相关的5个特征性模块和5个枢纽基因。其中black模块仅在pCR组观察到。black模块的GO富集表明放化疗敏感性可能与免疫应答及细胞吞噬的生物学过程有关。COMMD8为black模块筛选出的枢纽基因,在pCR组表达下调。COMMD8通过蛋白质的泛素化调控许多过程。其中最重要的是COMMD8与CCDC22结合,通过降解IκB增加NFκB释放[10]。NFκB是特异性和非特异性免疫反应的关键激活因子,调节细胞凋亡和细胞分裂并可诱导未成熟的树突状细胞向耐受性树突状细胞分化。Taher等将NFκB抑制剂作用于乳清蛋白过敏小鼠,发现嗜酸性粒细胞增多,IL-5和IL-13降低,IL-10升高,这表明更高的NFκB活性有助于诱导、提高机体耐受性[11-12]。Wang[13]等研究发现,COMMD8在非小细胞肺癌组织和细胞中异常上调,敲除COMMD8抑制了细胞增殖、集落形成和糖酵解,且加速了肿瘤细胞凋亡。COMMD8的下调是否增加了ESCC对放化疗的敏感性还有待进一步证实,但本研究结果为进一步阐明COMMD8在放化疗敏感性中的重要性提供了新的研究思路。
EPS15L1是Green模块中筛选出的枢纽基因。该模块仅在pCR组观察到。GO富集分析显示其参与了有丝分裂微管细胞骨架的形成。EPS15L1与其同源结构蛋白EPS15有相似的生物学功能,是EGFR的酪氨酸激酶底物,主要调控转铁蛋白受体的内吞作用,维持红细胞的发育和铁稳态。EGFR组成受体酪氨酸激酶(PTKs),PTKs的过渡表达与肿瘤增殖、迁移密切相关。EGFR及其信号组分已经被开发用于肿瘤新化疗药物的靶标,由于肿瘤的异质性和耐药性使得其有效性极大受限[14]。因此,EPS15L1作为EGFR上游调控物质有望成为新的治疗靶点。通过GO富集分析确定了Yellow模块参与了内皮细胞凋亡的调控。EPHB6为该模块筛选出的核心基因,是PTKs家族的一种缺乏酪氨酸激酶活性的特殊型受体。国内外多项研究表明,EPHB6在多种肿瘤组织中表达下调,并参与内皮细胞的增殖、迁移及重塑等过程[15-16]。在Toosi[15]等的研究中表明,高表达EPHB6的三阴乳腺癌细胞对阿霉素治疗更敏感,这说明EPHB6能将其转化为抵抗力弱的细胞,从而增加治疗的敏感性。本研究中与ESCC的nPCR组相比,pCR组的EPHB6高表达,与上述报道相符。因此,EPHB6有望成为肿瘤放化疗敏感性及判断预后的新靶点。
在本研究中,通过GO分析确定了blue模块为参与内质网组成和正向调控细胞生长的模块。MICAL2为blue模块的枢纽基因,是一种细胞骨架调控蛋白,在肾癌、胃癌、肺癌等多种肿瘤组织中均高表达,参与细胞的增殖、迁移、侵袭和上皮间质细胞转化。在nPCR组的患者中高表达,可认为是引起患者对放化疗不敏感的一个因素。Lu J[17]等的研究表明,MICAL2与P53结合,使P53留在细胞质中介导其泛素化,降低P53功能从而促进直肠癌的生长。王月媛[18]在对乳腺癌的迁移研究中发现,MICAL2可通过以Rac1依赖的方式抑制EGFR蛋白的降解,从而激活EGFR/P38通路从而促进乳腺癌细胞的迁移。此外,在对肺腺癌患者的研究中发现,MICAL2总体表达水平与淋巴转移及较短的生存期呈正相关[19]。基于这些结果,MICAL2可能是与ESCC放化疗敏感性相关的枢纽基因,其高表达使ESCC患者降低对放化疗的病理性反应。RBPMS是Red模块筛选出的枢纽基因,是RNA结合蛋白家族的成员,在mRNA的加工、运输及翻译中起重要作用。已有报道表明RBPMS家族能抑制AP-1信号通路,调节乳腺癌的增殖和迁移,并且通过调节Sox2的表达促进乳腺癌干细胞的自我更新及耐药[20]。
5个与放化疗敏感性相关的模块KEGG通路富集结果显示共有70条KEGG通路被富集。其中富集最多为PI3κ/Akt信号通路、cAMP信号通路和脂肪分解通路、氧化磷酸化通路。研究表明各种因素引起的PI3κ/Akt信号通路的激活会降低肿瘤对放化疗的敏感性。宁显会[21]等报道了NOX4通过激活PI3κ/Akt信号通路抑制鼻咽癌细胞对放疗的敏感性。钟伟杰[22]等研究指出,人骨髓来源间充质干细胞在肿瘤微环境中通过上调IL-17A激活PI3κ/Akt信号通路,增加了弥漫性大B细胞淋巴瘤的化疗耐药性。这可能与PI3κ/Akt信号通路调控细胞凋亡有关。
本研究利用WGCNA生物信息学方法,鉴定出与ESCC放化疗敏感性密切相关的基因模块和枢纽基因。MICAL2、EPHB6、COMMD8、EPS15L1、RBPMS是ESCC新辅助放化敏感性的关键基因,有望成为ESCC术前放化疗评估的分子标志物及潜在的治疗靶点。
