基于WGCNA研究激素性股骨头坏死相关核心靶点*
2021-05-25覃文涛蒋铭杨薄占东
覃文涛,胡 阳,蒋铭杨,薄占东
(广西医科大学第一附属医院,南宁 530021)
激素性股骨头坏死(steroid-induced avascular necrosis of the femoral head,SANFH)因其发病率和致残率高,已成为骨科常见且难治的疾病之一。近年来糖皮质等类固醇激素应用逐渐广泛,进一步的增加了SANFH 的发病率[1]。然而,SANFH 的发病机制一直以来都未有定论,多种治疗方法的疗效欠佳,因此,探索SANFH 的发病分子机制,已经迫在眉睫。加权基因共表达网络(weighted gene co-expression network analysis,WGCNA)是一种通过分析基因之间的相关性,将表达模式相似的基因分装在一个基因模块中,鉴定基因模块与表型的关系的一种网络分析方法[2-3]。可以根据模块与表型之间的相关性程度来鉴定生物标记物或者关键靶点。
本研究通过使用WGCNA 方法来鉴定早期SANFH 发生的潜在生物学标记物,从而提高早期SANFH的诊断治疗,降低致残率。
1 材料与方法
1.1 数据下载 从GEO(Gene Expression Omnibus,https//www.ncbi.nlm.nih.gov/gds)数据库中下载SANFH的芯片表达数据及临床信息(GSE123568),平台注释文件是Affymetrix 人基因表达阵列(GPL15207),GSE123568 数据集中包括10 例正常组和30例SANFH组表达数据。
1.2 筛选差异基因(DEGs)用R软件(版本3.6.3)对芯片数据集进行数据标准化处理和探针注释,用“limma”包进行样本差异分析[4-5],将差异倍数(FC)取以2 为底的对数,并以|Log2FC|>1,P<0.05 作为DEGs的筛选条件。
1.3 WGCNA 共表达模块分析 使用R 的“WGCNA”包构建SANFH相关的加权共表达网络[6],去掉表达量小于0.5 的样本,然后计算无尺度分布拓扑矩阵,利用“pickSoftThreshold”函数挑选出最佳软阈值β,并计算每个基因的皮尔森相关系数,以加权相关系数构建邻接矩阵,再将邻接矩阵转换为拓扑重叠矩阵,以此构建聚类树,将模块内基因数目大于50 的进行保留,相似度大于0.25 的模块进行合并,根据SANFH组和正常组的表达差异,选择最优模块。
1.4 功能富集分析 在WGCNA 最优模块中选择核心基因(R>0.8,P<0.01),通过与DEGs的交集得到交集基因,利用R 的“clusterProfiler”包对交集基因进行基因本体论(GO)和京都基因和基因组百科全书(KEGG)功能富集[7],取富集结果中P<0.05 和adjustP<0.05的前10个项目进行展示。
1.5 蛋白质互作网络(PPI)图构建及关键(Hub)基因选择 在线String 数据库(https://string-db.org/cgi/input.pl)可以预测蛋白质之间的相互作用关系[8],上传交集基因到String 数据库中,相互作用的最低阈值选择0.40,其他设置默认,导出结果文件。将得到的结果文件导入Cytoscape3.7.2中,按照高表达基因标记为红色,低表达设为蓝色,颜色深度表示差异大小来绘制PPI图,并使用cytoHubba对网络进行计算分析[9],寻找Hub基因。在网络中,分别计算出评价每个节点重要性的3 个指标:节点度(degree)、紧密度(closeness)和中心度(betweenness),筛选出排名前10的节点,它们共同的节点就是Hub基因。
1.6 Hub 基因功能及诊断性能分析R 的“cluster-Profiler”包完成对Hub基因的功能富集,挑选出P<0.05 的富集结果,用“plotROC“包对Hub 基因进行诊断性分析[10],绘制受试者工作特征曲线(ROC 曲线)。为探索Hub基因之间的相互联系,利用Gene-MANIA(http://genemania.org/)数据库线上分析[11-12],构建出Hub基因之间的互作网络图。
1.7 统计学方法 本研究所有数据采用R 软件分析,差异分析采用贝叶斯算法,相关性分析采用皮尔森相关性分析,两组间的数据分析采用双尾t检验,适当的时候用Welcht检验,以P<0.05为差异具有统计学意义。
2 结果
2.1 差异基因 根据GEO 数据库获得的40 例样本,经过差异分析,得到满足条件的384个DEGs,包括119 个上调基因,265 个下调基因,并以火山图和热图对DEGs进行展示,见图1。火山图中绿色表示下调,红色表示上调,热图显示DEGs 的表达在SANFH组和正常组中有明显差异。
2.2 WGCNA 和最优模块选择 用R 软件计算分析,确定了软阈值β=14,经过动态切割法得到分层聚类树,见图2A,共得到4个模块,模块特征的皮尔森相关性分析矩阵可以看出,turquoise模块与SANFH 的相关性最高(R=0.72,P<0.001),见图2B,以R>0.8,P<0.01 选取该模块的基因(n=375),并与DEGs取交集得到交集基因(n=225)。
2.3 交集基因GO 和KEGG 富集 利用R 的“clusterProfiler”包对225 个交集基因进行GO 和KEGG功能富集分析,见图3,GO富集结果显示,交集基因主要富集在红细胞发育、红细胞分化、骨髓细胞稳态、骨髓细胞发育和骨髓细胞分化等生物学功能(BP)上,细胞成份(CC)上主要在细胞器外膜、线粒体外膜和皮质细胞骨架有富集,此外还参与了分子功能(MF)的富集,包括泛素样蛋白转移酶活性、结合酶活性和细胞骨架的结构成分等。KEGG富集分析与线粒体、泛素介导的蛋白水解和导管酸分泌物等通路有关。
2.4 Hub 基因的PPI 构建 将225 个交集基因导入String 数据库,得到所有蛋白互作网络图,使用Cytoscape3.7.2 软件的cytoHubba 计算出degree、closeness 和betweenness 3 种节点打分排名前10 的关键基因,并找出所包含的共同的基因,这些基因就是Hub 基因,最终找到4 个Hub 基因,即GATA1、SLC4A1、EPB42 和DMTN,并构建了其关键基因的PPI图,见图4。

图1 差异基因的火山图和热图
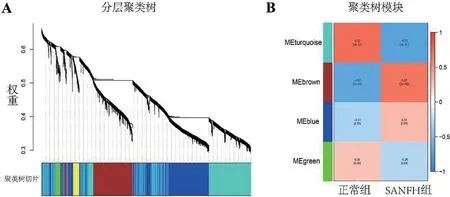
图2 WGCNA结果

图3 交集基因的GO和KEGG功能富集分析
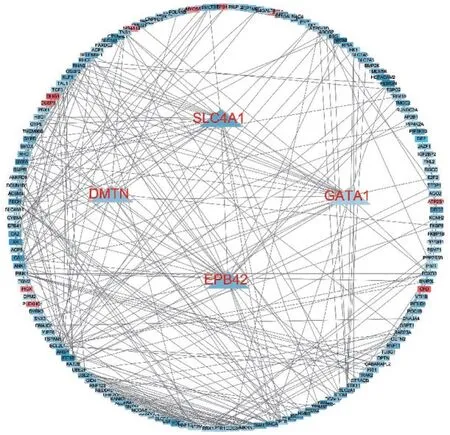
图4 蛋白网络互作图
2.5 Hub 基因的分析 每个Hub 基因在SANFH 中都有明显的差异表达,且表达都低于正常组,功能富集结果显示,Hub基因主要在凝血、红细胞和骨髓细胞发育、稳态及分化等分子生物学途径上有富集,见表1,这与DEGs 的富集结果有一致性。Hub基因的诊断性ROC 曲线显示,4 个基因在SANFH中具有很好的诊断价值,见图5A。基因之间的网络互作图也显示Hub基因之间联系紧密,见图5B。

表1 Hub基因的GO富集情况
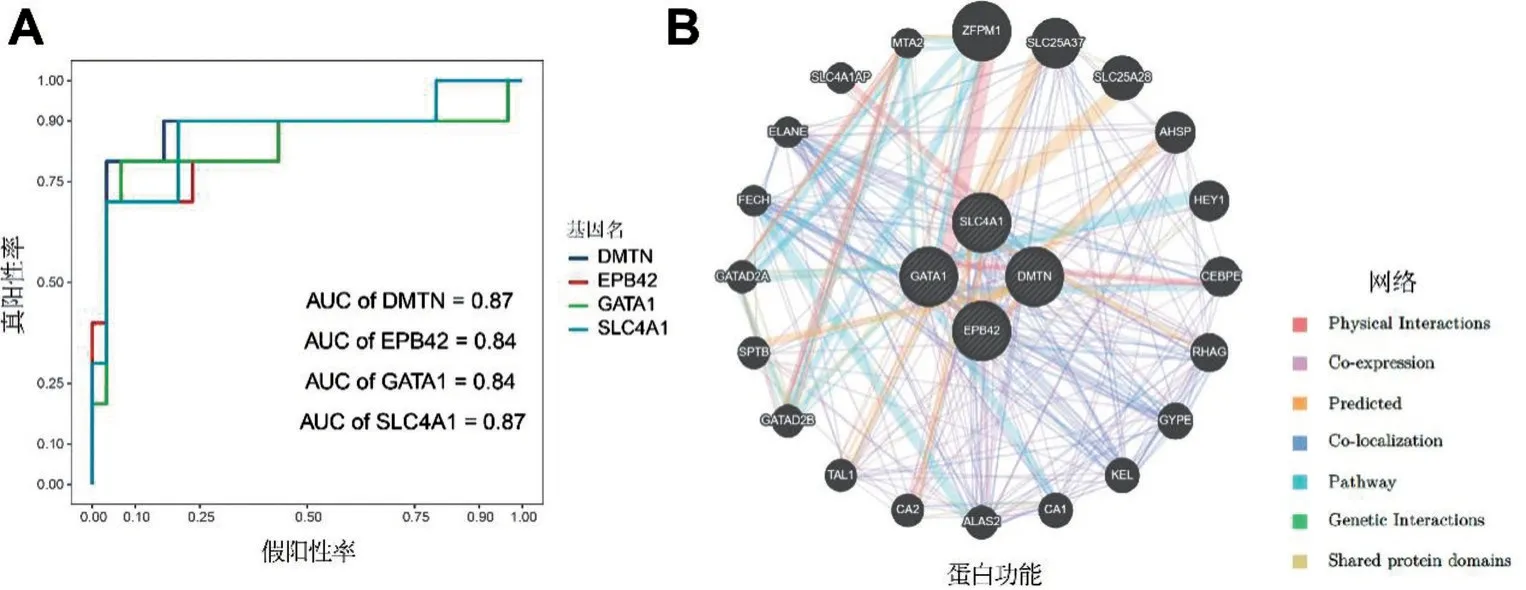
图5 Hub基因的诊断价值及其基因互作网络分析
3 讨论
SANFH是一种发病机制复杂,致残率极高的疾病,对中青年患者影响极大。截止目前,由类固醇激素引起的股骨头坏死依然是非创伤性股骨头坏死最常见的病因[13]。近年来,类固醇激素的大量应用导致SANFH 的患者逐年增加,为临床治疗工作带来极大的负担[14]。由于其发病机制尚不明确,目前很难找到针对患者早期的预防及治疗措施[15]。随着分子生物的发展,已有一批学者将目光投向基因层面的研究,在SANFH 的发病进程中基因的改变扮演着重要的角色[16-17],因此找到SANFH相关的致病基因,探明其具体的发生机制对临床SANFH 的早期诊断具有很重要的意义。
本研究通过对GEO 数据库中的GSE123568 芯片数据进行差异分析,找到正常组与SANFH 组间差异表达的基因,其中满足条件的差异基因共384个,其中上调的有119 个,下调的有265 个,下调基因明显多于上调基因,提示SANFH 发生时大部分基因的表达下调。通过WGCNA 挖掘出与SANFH相关性高的基因,将得到的结果与之前的差异基因取交集,得到同时具有差异性和相关性的交集基因。对相关度更高的交集基因进行GO和KEGG功能富集,发现这些基因主要富集在红细胞和骨髓细胞的发育、分化及稳态上。而有研究报道,骨髓基质细胞的凋亡是SANFH发生的主要特点之一[18],激素可以促进骨髓基质细胞向脂肪细胞细胞转化,抑制其向成骨细胞的方向的分化,从而导致骨坏死,进而发展为股骨头坏死[19]。同时有研究发现,促红细胞生成素可以通过刺激网织红细胞的增值和分化,促进血管生成来预防类固醇激素引起的股骨头坏死发生[20]。这些研究与交集基因富集的结果有较好的一致性,提示在交集基因中存在对股骨头坏死发生和发展起到关键作用的基因。
通过Cytoscape3.7.2软件的相关节点算法分析,最后从交集基因中找到了4 个Hub 基因:GATA1、SLC4A1、EPB42 和DMTN,这可能是SANFH 发生的关键基因。4 个Hub 基因在正常组和SANFH 组中的表达差异明显,在SNAFH 组中均出现低表达。从富集的结果可以看出,Hub 基因的富集结果与384 个差异基因的富集结果具有一致性,进一步证实了这4 个基因的关键作用。GATA1 是类红细胞发育的转录激活或者抑制因子,有研究显示,GATA1 在低氧环境中可以诱导红系造血[21],而在早期SANFH 中,缺血缺氧是骨坏死中的重要表现[22],使用一定量的糖皮质激素后可以阻碍股骨头内的血液供应[23],从而形成缺血缺氧的环境。本研究中,GATA1 在SANFH 中表达降低,可能造成股骨头缺氧加重。SLC4A1、EPB42 和DMTN 3 者具有类似的功能,均在维持红细胞正常的形态和功能上具有重要的作用[24-26],他们在SANFH中表达含量也明显下降,可能导致股骨头内正常红细胞含量降低,造成股骨头内缺血缺氧。因此,从4 个Hub 基因的相关性研究中,表明低表达的Hub 基因是SANFH 发展中的危险因素,在SANFH 的早期发病中具有重要的研究价值。对经常需要服用激素的患者,及时检测血液中4 个Hub 基因的表达,将会一定程度上提高早期SANFH 的诊断率和治疗效果,从而降低致残率。
综上所述,本研究采用生物信息学分析的方法,发现4个Hub基因在SANFH中的表达具有显著差异,低表达的Hub基因可能通过参与缺氧信号通路来促进SANFH 的发展,为临床SANFH 的早期诊断和治疗提供了可靠的分子标记,同时也为今后探明SANFH的发病机制提供了一个参考方向。
