苹果坏死花叶病毒实时荧光定量PCR 检测体系的建立*
2021-05-24胡国君张尊平范旭东马勉娣张秀英鲁兴凯董雅凤
胡国君,张尊平,范旭东,任 芳,马勉娣,张秀英,鲁兴凯,董雅凤
(1 中国农业科学院果树研究所,国家落叶果树脱毒中心,辽宁兴城 125100)(2 昭通市苹果产业发展中心)
花叶病是苹果上的一类重要病毒病害,发生普遍,危害严重,典型症状为叶片上形成大小不一的鲜黄色或黄白色斑块。发病后苹果叶片叶绿素总量可降低14.80%~38.72%、净光合速率降低 2.93%~45.83%,果实减产30%~50%[1-2]。20 世纪30 年代该病害在欧洲首次报道[3],其病原为苹果花叶病毒(apple mosaic virus,ApMV),我国最早的文献记载在20 世纪50 年代末[4]。目前,我国关于该病害的研究主要集中在症状观察及发生情况调查等方面,深入研究较少,根本原因是作为病原的ApMV 在我国检测比较困难[5]。2017 年,日本学者在从我国引进的表现花叶症状的苹果树上检测到1 种雀麦花叶病毒科(Bromoviridae)等轴不稳环斑病毒属(Ilarvirus)的新病毒,并命名为苹果坏死花叶病毒(apple necrotic mosaic virus,ApNMV)[6]。经进一步研究确认,ApNMV 为我国苹果花叶病的主要病原[7],在韩国和印度的苹果树上也发现了该病毒[8-9]。据报道,山楂也是ApNMV 的寄主[10]。ApNMV 为正义单链RNA 病毒,由3 条链组成(RNA1、RNA2和RNA3)。RNA1 和RNA2 编码2 个聚合蛋白,与病毒的复制相关;RNA3 编码运动蛋白和外壳蛋白,分别位于基因组的5′和3′端[6]。
目前,苹果病毒主要以核酸检测为主,如常规PCR、巢式PCR、多重PCR 和实时荧光定量PCR(quantitative real-time PCR,qPCR)等。其中,qPCR作为一种灵敏度高、特异性和可靠性强、稳定性好的检测技术,已被广泛应用于果树病毒检测[11-13]。由于qPCR 是在封闭的环境下进行反应,通过收集荧光信号来判断反应结果,与巢式PCR 等同样灵敏度较高的检测方法相比,具有数据读取简单、不易产生污染等优点。邢飞[14]建立了 ApNMV 一步RT-qPCR 探针检测方法。本研究建立了一种ApNMV 的TB Green 染料法qPCR 检测方法,可对田间不同生长周期不同组织部位的苹果样品进行检测,有效提高了ApNMV 的检测效率。
1 材料与方法
1.1 试验材料
以实验室保存的8 株携带ApNMV 的苹果树作为qPCR 引物筛选的测试样品,其中,‘富士’和‘响富’各2 株,‘华红’‘天宏’‘嘎拉’、晋18 各1 株,以无毒砧木组培苗‘B9’为阴性对照。其余检测样品均采集于中国农业科学院果树研究所的试验田。
1.2 试剂及仪器耗材
Taq™酶、10×PCR Buffer(Mg2+plus)、dNTPs、PrimeScript™ RT reagent Kit with gDNA Eraser Perfect Real Time、TB Green®Premix Ex Taq™(Tli RNaseH Plus)和大肠杆菌感受态细胞DH5α,购自宝日医生物技术(北京)有限公司(TaKaRa);胶回收试剂盒购自爱思进公司(Axygen);pTOPO-TA Vector 购自北京艾德莱生物科技有限公司;qPCR用耗材Optical Flat 8-Cap Strips for 0.2 mL tube strips/plates 和Low-Profile PCR Tubes 8-tube strip(white)、仪器CFX Connect™ Real-Time System,购自伯乐生命医学产品(上海)有限公司(Bio-Rad);反转录酶M-MLV 购自普洛麦格公司(Promega);DNA Mark II 购自北京天根生化科技有限公司;DNase/RNase Free dH2O 购自生工生物工程(上海)股份有限公司;超微量紫外可见分光光度计(DS-11)购自美国丹诺尔公司(Denovix)。
1.3 总RNA 提取及cDNA 合成
取苹果枝条韧皮部或叶片样品50 mg,采用吸附柱法[15]进行总RNA 提取,利用超微量紫外可见分光光度计(DS-11)进行总RNA 浓度测定及质量分析,-80 ℃保存备用。采用去除基因组DNA 反转录试剂盒(PrimeScript™ RT reagent Kit with gDNA Eraser Perfect Real Time,Takara)合成cDNA:取5×gDNA Eraser buffer 2.0 μL、gDNA Eraser 1.0 μL 及总 RNA 1 μg,加RNase Free ddH2O 至总体积为10 μL,混匀,42.0 ℃ 2 min,放冰上。然后加入含PrimeScript RT Enzyme MixⅠ1.0 μL、RT Primer Mix 1.0 μL、5×PrimeScript buffer 2 4.0 μL 和RNase Free ddH2O 4.0 μL 的混合液,混匀,37 ℃ 15 min,85 ℃5 s,产物在冰上冷却后-20 ℃保存备用。
1.4 引物设计、鉴定及筛选
分别根据GenBank已登录的ApNMV 3条RNA链上的保守序列,设计了12 对ApNMV qPCR 检测引物(表1),由生工生物工程(上海)股份有限公司合成。
采用常规PCR 对引物的特异性进行筛选。反应体系为25 μL,含10×PCR buffer(Mg2+plus)2.5 μL、dNTP Mixture(2.5 mmol/L each)0.5 μL、10 μmol/L正向、反向引物各0.5 μL、5 U/μL rTaq® 0.125 μL和 cDNA 2 μL,用dH2O 补足至25 μL。反应条件:94 ℃ 3 min,94 ℃ 15 s,55 ℃ 15 s,72 ℃ 20 s,35 个循环;72 ℃延伸7 min。扩增产物用1.2%琼脂糖凝胶电泳检测。
采用qPCR 对引物的检测效果进行筛选。反应体系为20 μL,含TB GreenPremix Ex TaqII(Tli RNaseH Plus)(2X)10.0 μL、10 μmol/L 正、反向引物各0.5 μL、DNase/RNase Free dH2O 7 μL 和cDNA 2 μL。扩增条件:94 ℃预变性32 min,94 ℃10 s,58 ℃ 15 s,72 ℃ 15 s,40 个循环,延伸结束时记录荧光信号。熔解曲线分析的温度范围是60~95 ℃,每5 s 增加0.5 ℃,以鉴别引物二聚体和非特异性扩增。
PCR 扩增产物经回收纯化后与 pTOPO-TA Vector 连接,转化大肠杆菌感受态细胞DH5α。挑取白色菌落进行PCR 鉴定,将阳性克隆送北京诺赛基因组研究中心有限公司测序。将获得的序列在GenBank 数据库中进行同源性分析。
以‘华红’样品100~10-7梯度稀释的cDNA 为模板,构建qPCR 各引物的标准曲线。

表1 ApNMV 实时荧光定量PCR 引物
1.5 qPCR 扩增条件的优化
将感染ApNMV 的‘华红’样品总RNA 反转录合成的cDNA 作为模板,分别采用终浓度为200、300、400、500 nmol/L 的引物进行qPCR 扩增,根据Cq值和扩增效果筛选出每对引物的最佳浓度。在最佳引物浓度下,检测不同引物在退火温度分别为52.0、54.0、55.9、58.4、60.3、62.0 ℃时荧光信号的变化情况,选取扩增效果最好时的退火温度。
1.6 qPCR 检测的灵敏度
以‘华红’样品100~10-7梯度稀释的cDNA 为模板,分别进行qPCR 和常规PCR 扩增,比较不同方法的灵敏度。
1.7 田间样品检测
2019 年12 月至2020 年1 月在田间从休眠苹果枝条上采集60 个样品(9 个样品叶片在夏季表现花叶的症状),分别采用常规PCR 和qPCR 检测ApNMV,比较2 种方法的检测效果。
2 结果与分析
2.1 引物的筛选及鉴定
以无毒砧木‘B9’试管苗为阴性对照,选取8个ApNMV 阳性苹果样品进行常规PCR 检测。12对引物在所有阳性样品中均扩增到目标片段大小的条带(图1),阴性对照和未加模板的空白对照未扩增到条带,但引物ApN2-F3/R3、ApN1-CP-F1/R1和ApN1-CP-F2/R2 的扩增产物不是单一条带,有杂带,引物ApN1-F2/R2 的二聚体比较明显,引物利用率低。回收其余8 对引物扩增‘华红’苹果样品的PCR 产物,进行克隆转化并送测序。将所获得的序列在NCBI 数据库上进行Blast 分析,结果显示,ApN1-MP-F2/R2 的扩增序列不是ApNMV 序列,其余7 对引物的扩增序列均为ApNMV 特异序列,与GenBank 中已登录的ApNMV 分离物的核苷酸序列同源性为93.6%~98.8%。
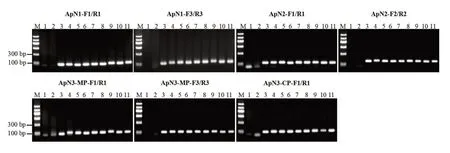
图1 7 对ApNMV 引物常规PCR 扩增产物的电泳图
选取ApNMV 阳性苹果样品‘华红’和‘响富’及阴性苹果样品‘B9’进行qPCR 检测。提取的3个样品的总RNA 浓度分别为420.86、441.59、407.73 ng/L,3 对引物对3 个样品的扩增曲线Cq值分别为ApN1-F3/R3(18.90、18.53 和34.49)、ApN2-F1/R1(17.42、17.61 和34.93)和ApN3-MP-F3/R3(17.59、17.48 和34.79)。内参引物EF-1α-F/R 对2 个样品的扩增曲线Cq值分别为15.36 和15.16,且熔解曲线为单一峰。
以‘华红’样品100~10-7梯度稀释的cDNA 为模板,分别构建3 对ApNMV 特异引物的标准曲线。由表2 可知,引物ApN1-F3/R3 的扩增效率最高。
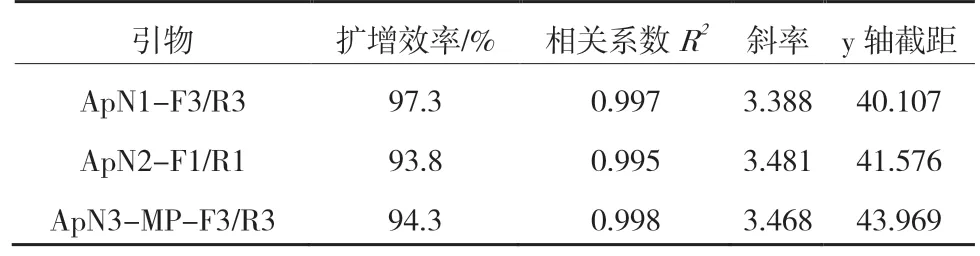
表2 各ApNMV 引物标准曲线的扩增效率、相关系数、斜率、截距
2.2 qPCR 反应体系优化
在52.0~62.0 ℃间设置了6 个退火温度,qPCR扩增Cq值为17.35~17.62,不同温度间的Cq值差异不明显,仅为0.27,当退火温度为60.3 ℃时,相对荧光强度相对较强,且熔解曲线为单一峰,作为最佳退火温度(图2-A)。在60.3 ℃下,4 个引物浓度间的Cq值为17.03~18.12,不同浓度间的Cq值差异较大,其中引物浓度为300 nmol/L 时,与其他3 个浓度区分明显,Cq值最小,为17.03(图2-B),因此,将300 nmol/L 作为最终引物浓度。

图2 不同退火温度(A)和引物浓度(B)下ApNMV 的qPCR 扩增结果
2.3 qPCR 检测的灵敏度
qPCR 和常规PCR 检测灵敏度测试结果表明,常规PCR 能检测到稀释倍数为10-3的ApNMV 阳性苹果样品的cDNA(图3-A),而qPCR 可检测稀释倍数为10-5的ApNMV 阳性苹果样品的cDNA,此时Cq值为33.78,仍小于阴性对照的Cq值(34.49),qPCR 检测灵敏度比常规PCR 检测提高了100 倍(图3-B)。

图3 不同稀释梯度苹果样品常规 PCR(A)和qPCR(B)检测灵敏度比较
2.4 田间样品检测
应用常规PCR 和所建立的qPCR 方法对60 个苹果样品进行ApNMV 检测,检测部位为枝条韧皮部,常规PCR 的检出率为51.7%,qPCR 的检出率为56.7%。2 种方法在9 个有花叶症状的苹果样品中均检测到ApNMV;在未表现症状的苹果样品中,qPCR 检测到ApNMV 的数量比常规PCR 多检测到3个。
3 结论与讨论
病毒病已然成为危害我国苹果产业的重要病害,按症状表现可分为潜隐性和非潜隐性病害2 大类型。花叶病是重要的显症病毒病害,在我国各个苹果产区均有发生[7,17]。病毒病不能通过化学药剂进行防治,栽植无病毒苗木是最有效的防控措施,而建立灵敏、可靠、快速的病毒检测技术可为病毒病防控提供重要的技术保障,同时对苹果苗木和种质资源的认证及检疫工作也具有重要意义。受病原的限制,我国对苹果花叶病的检测技术研究还比较少。
本研究针对已报道的ApNMV 分离物3 条RNA链的保守区域设计12 对引物,分别进行常规PCR扩增和目标片段的克隆测序,通过扩增效果比较及序列比对分析,确定7 对ApNMV 特异引物;进一步比较7 对引物对2 个阳性样品扩增曲线的Cq值,分别从3 条链上各筛选出1 对可用于qPCR 检测引物;最后通过构建标准曲线,获得3 对引物的扩增效率,确定引物ApN1-F3/R3 用于ApNMV 的qPCR分析。通过引物浓度和退火温度的优化,建立了以ApN1-F3/R3 为引物的qPCR 检测方法,可特异性检测ApNMV,大大提高了检测效率,灵敏度是常规PCR 检测的100 倍。该检测方法适用范围广,对不同生长时期和不同部位的苹果样品均有良好的检测效果。
引物的特异性、病毒在样品中的含量和取样时间是影响以核酸为基础的病毒检测方法的关键因素[18]。此外,病毒群体的遗传多样性也影响检测效率,病毒基因组高度变异可直接导致检测失败或假阴性出现[12]。与其他方法相比,qPCR 无需电泳凝胶和染色,节省了病毒检测的时间,并具有特异性强、灵敏度高、病毒可以被量化等优点,能够满足针对各种类型的植物样品中ApNMV 的检测和带毒量测定。本研究为苹果脱毒苗提供了有效的认证检测方法,也为ApNMV 的流行监测与种群生态学研究提供了支撑。
