CRISPR-Cas系统编辑丝状真菌的进展与挑战
2021-05-22肖晗刘宜欣
肖晗,刘宜欣
(上海交通大学生命科学技术学院,微生物代谢国家重点实验室,教育部代谢与发育科学国际合作联合实验室,上海200240)
丝状真菌大多隶属于子囊菌和担子菌,是一类对生物、环境和材料等学科有着重要意义的微生物。丝状真菌是蛋白分泌的理想宿主,其中曲霉属和木霉属的菌株被广泛用于生产木聚糖酶和纤维素酶[1]。不少担子菌因具有较强的重金属吸附能力,在环境污染治理方面展示出较大潜力[2]。更为重要的是,丝状真菌能天然合成多种具有重要生物学活性的次级代谢物,例如青霉菌生产的青霉素[3]、曲霉生产的洛伐他汀[4]和灵芝生产的灵芝酸[5]等等,是生产高附加值化合物的细胞工厂。
基于丝状真菌的各项基础和应用研究高度依赖基因编辑平台,比如鉴定基因功能[6]、激活沉默基因簇[7]等。然而,丝状真菌独具的生理性能给这类微生物基因编辑平台的构建带来挑战。这主要表现在以下四方面:
(1)顶端生长 丝状真菌通过顶端菌丝体的分裂实现生长,外源质粒难以均匀分布到分裂的多个菌丝体中[8]。菌丝体的胞质成分甚至细胞器可通过隔膜孔进入到相邻细胞中,造成抗性筛选的假阳性。
(2)异核性 担子菌中的菌丝体细胞通常包含2个不同的细胞核,如果包含有抗性筛选标记的转化子没有同时整合到两个核中,后续的转化子由于分裂稀释会不再包含抗性标记[8]。
(3)同源重组效率低 同源重组是精准基因编辑的基础,大部分丝状真菌中非同源性末端接合(non-homologous end joining, NHEJ)的修复机制占主导地位,很难实现基于同源重组的精准基因编辑[9]。
(4)遗传筛选标记匮乏 由于遗传操作工具不成熟,营养缺陷型标记在不少丝状真菌中尚不可用,可用的筛选标记仅有少数几种抗生素(如萎锈灵[10]、潮霉素[8]等)的抗性基因。
近年来,基于RNA介导的核酸内切酶CRISPRCas 系统能在基因组特定位点引入DNA 或RNA 的链缺口,诱发宿主启动自身的防御机制修补缺口,大幅提高了同源重组效率,减少对筛选标记的依赖,实现多个物种的多基因编辑[11-13]。随着研究的深入,CRISPR-Cas 系统也被应用于丝状真菌中,为构建成熟的丝状真菌基因编辑平台奠定基础。本综述重点介绍了近三年丝状真菌基因编辑的最新进展,讨论了该系统协助丝状真菌基因编辑存在的挑战及可能的解决方法。
1 CRISPR-Cas 系统编辑丝状真菌的进展
自2015 年Liu 等首次将CRISPR-Cas 系统引入模式丝状真菌里氏木霉中以来[14],5 年多时间里,该系统在越来越多的丝状真菌中成功构建,它不但能更加高效、快速地编辑模式菌株,更为重要的是,它协助很多传统方法无法实现基因编辑的菌株进行基因编辑[9,15],大幅提高了人们对这类重要微生物的认识和改造。本部分围绕CRISPRCas 系统的递送、体内表达、同源臂设计和宿主改造几方面介绍了其编辑丝状真菌的进展。
1.1 CRISPR-Cas系统的递送
聚乙二醇(polyethylene glycerol,PEG)和根瘤农杆菌介导的原生质体转化(agrobacteriummediated transformation,AMT)是把CRISPR-Cas系统递送到丝状真菌中最主要的两种方式[16]。外源DNA 片段经PEG 介导的转化后,通常以多拷贝的形式随机整合到丝状真菌基因组上[17],而AMT则倾向于将外源片段以单拷贝的形式随机整合到基因组上[18]。经PEG 或AMT 递送的CRISPR-Cas系统,可以是DNA、RNA[9,19],也可以是体外表达的Cas 蛋白和体外转录的引导RNA(guide RNA,gRNA)组装好的CRISPR-Cas 核糖核蛋白(ribonucleoprotein,RNP)[20-22]。由于提供了一种瞬时的、不依赖于体内表达元件和遗传筛选标记的基因编辑方式,RNP在丝状真菌中的应用得到越来越多的关注。在2019年的一项研究中,Li等[23]开发了一种仿生矿化的RNP 递送系统,将利用磷酸钙矿化的纳米颗粒承载RNP 递送至植物病原真菌稻瘟病菌的原生质体中。仿生矿化处理的RNP 可保护gRNA 免受酶水解,提高RNA 的稳定性,保留Cas9蛋白的酶活性。同时,他们还使用聚丙烯酸改善仿生矿化的RNP 纳米颗粒(biomimetic mineralized RNP nanoparticle,BM-RNP NP)的分散性。通过观察与BM-RNP NP 有相似形态和尺寸分布的EGFP 纳米颗粒(BM-EGFP NP)的递送过程,发现BM-EGFP NP 附着在细胞膜表面可能会增加经胞吞途径被细胞摄取的概率。研究者将中断编码Sytalone dehydratase 的sdh 基因的BM-RNP NP 加入原生质体中,检测发现该基因的编辑效率达20%,而仅通过RNP 处理的细胞中该基因的表达并未发生明显变化。进一步序列分析显示,用BM-RNP NP 处理的米曲霉在7 个潜在的脱靶位点均未发生变化。他们认为使用仿生矿化的RNP 进行基因编辑有望提供一种通用且不依赖DNA 的方法来提高编辑效率。
此外,CRISPR-Cas 系统还能介导外源片段在丝状真菌中的定向整合,彻底改变外源片段随机整合的递送模式。Lu 等[24]在根瘤农杆菌介导的TDNA 中间插入了靶向mfa2 基因的CRISPR-Cas 表达盒,在潮霉素筛选条件下,原本随机插入的TDNA片段能定向插入到经CRISPR-Cas切割的mfa2的靶位点,实现该基因的中断。更进一步地,将CRISPR-Cas 系统靶向到上述插入至mfa2 的CRISPR-Cas 表达盒的潮霉素抗性基因,可在潮霉素抗性基因的靶位点插入同义突变的mfa2 基因用以规避CRISPR-Cas 的识别,实现该基因的回补。类似地,通过巧妙设计CRISPR-Cas 系统,可将外源片段定向整合到基因组的特定位点。Sarkari等[25]首先利用CRISPR-Cas 系统破坏了黑曲霉(Aspergillus niger)整合靶位点pyrG 基因,使得突变株成为尿嘧啶营养缺陷菌株。然后,在含有递送外源片段的表达盒两端引入回复pyrG 功能的同源臂,在同源臂的两端再引入另一段靶向pyrG 的识别序列。基于此,在体内产生功能的CRISPR 系统将切割三处识别序列,切割含有外源片段质粒的两处后产生线性化的、与pyrG 具有同源臂的外源片段,该片段可定向插入到第三处切割pyrG 位点产生的双链缺口处,此时pyrG功能得到回复。
1.2 CRISPR-Cas系统的体内表达
在丝状真菌中构建基于CRISPR-Cas 的基因编辑系统的关键在于让该系统在丝状真菌中工作,即执行对目标DNA/RNA 的切割或靶向的功能。和体外表达CRISPR-Cas 系统相比,在丝状真菌体内表达CRISPR-Cas 系统具有不可替代的优势,也是CRISPR-Cas 系统在丝状真菌中研究最多、发展最快的方向之一。其原因可能有两方面:一方面是因为持续表达的CRISPR-Cas 能提高对目标DNA/RNA 的切割或靶向的概率,进而提高基因编辑效率;另一方面,在表达Cas 蛋白的菌株中递送gRNA 可降低Cas 蛋白和gRNA 无法进入同一细胞核的风险。本小节将系统介绍Cas 蛋白和gRNA 在丝状真菌体内表达的进展。
1.2.1 Cas蛋白的表达
Cas 蛋白的密码子优化被认为是促使它在丝状真菌中发挥功能的关键策略。Liu 等[14]尝试用人源密码子优化的Cas9 在丝状真菌里氏木霉(Trichoderma reesei)中构建CRISPR-Cas 系统时并未获得成功,采用经里氏木霉的密码子优化的Cas9 成功构建了CRISPR-Cas9 系统。除了里氏木霉外,采用特定宿主密码子优化Cas9 的策略在多种丝状真菌如稻瘟病菌(Pyricularia oryzae)[26]、灵芝(Ganoderma lucidum)[9]等中,也成功构建了基于CRISPR-Cas 的基因编辑系统。为了保证Cas 蛋白在丝状真菌的高效表达,除了选用内源强启动子外[27],最新一项研究发现在Cas 蛋白中引入内含子也能提高灵芝的基因编辑效率[28]。在Cas9 基因的上游引入灵芝内源3-磷酸甘油醛脱氢酶编码基因gpd 5'端的内含子可使得CRISPR-Cas9中断5'-单磷酸脱羧酶编码基因ura3 的效率提高10.6 倍,每转化107个原生质体可获得14~18 个基因编辑的突变株,研究者推测这可能是因为内含子的加入有利于异源基因的mRNA 在担子菌中的积累,进一步增强蛋白的表达[28]。来源于构巢曲霉(Aspergillus nidulans)的AMA1 序列是迄今为止发现的唯一能在曲霉属中自主复制的元件,Katayama等[29]利用截短至一半的AMA1 序列驱动CRISPR 质粒的复制,将多株米曲霉(Aspergillus oryzae)的基因编辑效率提高到50%~100%,这可能由于AMA1 在细胞以多拷贝的形式存在,提高了Cas9 和gRNA 的表达所致。在该质粒上加入了能抑制菌丝体生长的aoace2 基因表达盒,诱导表达该基因使得CRISPR 质粒快速丢失,便于多轮的基因编辑[29]。
另外,诱导表达的Cas9 由于可调控CRISPRCas系统基因编辑时的活力,减少脱靶效应等,在丝状真菌中也有不少应用。Weber等[30]利用一个合成的四环素依赖的系统诱导Cas9 在烟曲霉(Aspergillus fumigatus)中表达,在转化前加入四环素筛选表达Cas9 的菌株,将gRNA 表达盒和分裂失活的筛选标记整合到基因组上,该分裂的筛选标记中间有CRISPR-Cas 系统的靶向目标基因的识别序列,两侧是自我同源序列,经切割后同源重组回复筛选标记的基因功能。通过该设计,可以经筛选获得靶基因发生编辑的突变体。
基于CRISPR-Cas系统的靶向功能,Huang等[31]在黑曲霉(A. niger)中开发了单碱基编辑器。具体而言,他们将不具有切割DNA 活力的nCas9 或dCas9 与大鼠的胞嘧啶脱氨酶rAPOBEC1 融合,把CRISPR-Cas 识别的靶序列的胞嘧啶转化为胸腺嘧啶(图1)。该编辑器编辑报告基因pyrG 和fwnA 的效率为47.36%~100%,编辑非报告基因prtT 的效率为60%,当胞嘧啶位于CRISPR-Cas 识别序列的第2到第9个位置之间时,均可被编辑。
虽然Cas9至今仍是丝状真菌中应用最多的Cas蛋白,但近两年,开发Cas12a(Cpf1)在丝状真菌中进行基因编辑的研究逐渐增多[32-35]。Cas12a和Cas9 同属于Class ⅡCRISPR-Cas 系统,识别序列、切割方式并不相同。Cas12a 识别的原间隔序列(protospacer)为5'端富含胸腺嘧啶的TTTN,而Cas9 识别的protospacer 为3'端富含鸟嘌呤的NGG;Cas12a 切割DNA 双链产生黏性末端,而Cas9 切割双链DNA 产生平末端。和Cas9 相比,Cas12a 可以识别一个简单的转录本中多个串联的crRNA,为多基因编辑提供便利。Liu 等利用密码子优化的AsCpf1 在嗜热毁丝菌(Myceliophthora thermophila)中构建了基于CRISPR-Cas12a的多基因编辑系统。他们用一个单独的crRNA 阵列或三个单独的crRNA 同时靶向碳代谢抑制的转录因子cre-1 和两个与纤维素酶生产相关的基因res-1 和gh1-1,在一步转化中除了包括Cas12a 和crRNA 的库(或单一阵列)以外,还有供体DNA 用于修复缺口。和只转化供体DNA 的对照相比,用crRNA库和crRNA 阵列分别达到40%和32%的三基因编辑效率。基于CRISPR-Cas12a 介导的高效多基因编辑,Liu 等[35]进一步开发了CRISPR-Cas 辅助的标记回收技术。在编辑的多基因的其中一个基因内部插入neo 标记,在第二次编辑其他基因时同时靶向neo将其敲除,同时引入bar标记。以此类推,在连续转化中迭代替换标记基因,减少对多个筛选标记的依赖。此外,研究者们还在构巢曲霉(A.nidulans)[34]、棉阿舒囊霉菌(Ashbya gossypii)[33]等丝状真菌中建立了基于CRISPR-Cas12a 的基因编辑工具。在最新的一项研究中,将不具有切割DNA活力的dCas12a 和一个含有三个转录激活结构域的转录激活因子VP64-p65-Rta(VPR)融合(图1),靶向调节构巢曲霉(A.nidulans)中生物合成基因簇的调控区域,可激活基因表达并提高天然产物的产量[32]。
1.2.2 gRNA的表达
作为CRISPR-Cas 系统中另一核心组成成分,gRNA 的高效表达对于该系统在丝状真菌中功能实现也同样重要。gRNA 在真核细胞中的表达是由RNA 聚合酶Ⅲ型启动子驱动的,在对体内RNA 聚合酶Ⅲ型启动子元件不甚清楚的时候,不少研究者采用了转化经体外转录的成熟的gRNA 的方式,成功实现了黑曲霉(A.niger)[36]、里氏木霉(T.reesei)[14]、灵芝(G. lucidum)[9]、多节孢属菌(Sporormiella minima)[37]等多种丝状真菌的基因编辑。但是,和体内转录gRNA 相比,体外转录gRNA 虽然不依赖体内的启动子元件转录,也存在以下局限:①成本高,过程复杂;②很难保证细胞对其完全吸收;③进入胞内后,在等待Cas蛋白表达的过程中,可能被胞内RNA 酶降解而进一步降低RNP 的浓度。此外,还有研究认为体外转录的gRNA 由于稳定性较差不适合转化丝状真菌[38]。
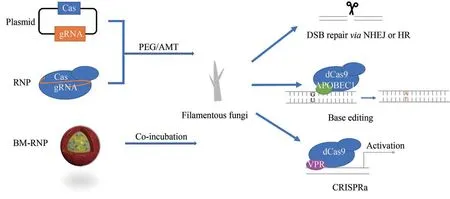
图1 CRISPR-Cas系统介导的丝状真菌基因编辑[RNP—核糖核蛋白;BM-RNP—仿生矿化的RNP;PEG—聚乙二醇;AMT—根瘤农杆菌介导的原生质体转化;DSB—DNA 双链缺口;NHEJ—非同源末端连接;HR—同源重组;APOBEC1—载脂蛋白B mRNA 编辑酶(胞嘧啶脱氨酶);VPR—含有三个转录激活结构域的转录激活因子;CRISPRa—CRISPR介导的基因转录激活系统]Fig.1 CRISPR-Cas system assisted gene editing for filamentous fungi(RNP—ribonucleoprotein; BM-RNP—biomimetic mineralized RNP; PEG—polyethylene glycerol; AMT—agrobacterium-mediated transformation;DSB—double stranded break;NHEJ—non-homologous end joining;HR—homologous recombination;APOBEC1—apolipoprotein B mRNA editing enzyme;VPR—VP64-p65-Rta,a tripartite transcriptional activator domain;CRISPRa—CRISPR activation)
不难看出,体内转录gRNA的方法往往更受欢迎。在丝状真菌中体内表达gRNA主要分为两类元件:一类是用RNA 聚合酶Ⅱ型启动子和具有自我剪切功能的核酶;另一类是用宿主内源的RNA 聚合酶Ⅲ型启动子(图2)。真核生物中RNA 聚合酶Ⅱ型启动子比Ⅲ型更为常见。但RNA 聚合酶Ⅱ型启动子负责mRNA 的转录,转录后会在转录本5'端加帽,3'端加PolyA 尾,引导转录本进入细胞质进行蛋白翻译。在gRNA 的5'端引入锤头型核酶HH和3'端引入丁型肝炎病毒(HDV)核酶将介导转录本自我剪切去掉两端的修饰,避免后续出核而无法行使引导和靶向的功能。利用该方法驱动gRNA 的表达在棘孢曲霉(Aspergillus aculeatus)[39]、互生交链孢菌(Alternaria alternata)[40]、皮炎芽生菌(Blastomyces dermatitidis)[41]、黑曲霉(A.niger)[42]等丝状真菌中实现了基因编辑。关于利用宿主内源的RNA 聚合酶Ⅲ型启动子驱动gRNA的表达,u6 基因的启动子在丝状真菌中使用最广泛[15,43-46]。我们在最新一项研究中发现,利用灵芝u6 基因的启动子驱动gRNA 转录时,在3'端引入HDV 能进一步提升基因编辑的效率,推测可能是因为由PolyT 终止的转录本会产生不同长短的含有T 的gRNA,而引入HDV 后产生了大小均一的gRNA,有利于RNP 更好地发挥功能[15]。除了U6启动子外,5S rRNA基因的启动子能更加高效驱动gRNA 在黑曲霉中的表达。Zheng等[47]用5S rRNA基因启动子表达gRNA,中断黑曲霉(A. niger)的一个假定的多肽合成酶编码基因albA 的效率高达96%,而用不同的U6 启动子中断albA 的效率只有15%~23%。他们还发现利用截短5'端区域的5S rRNA 基因启动子并没有明显降低CRISPR-Cas的编辑效率。此外,Song 等[48]测试了37 个tRNA启动子驱动CRISPR-Cas 系统在黑曲霉中基因编辑的效率,发现其中36 个都能成功介导目标基因的编辑[48]。在2019 年的一项研究中,Shi 等[38]系统比较了三种体内表达gRNA的方式对水稻恶苗病菌基因编辑效率的影响[38]。当用RNA 聚合酶Ⅱ型启动子加上自我剪切核酶的方式中断Fusarium cyclin C1 编码基因fcc1 时,并不能获得基因编辑的突变体。而用RNA 聚合酶Ⅲ型U6和5S rRNA 启动子中断fcc1 时,效率分别是37.5%和79.2%,表明用5S rRNA启动子表达的策略更为有效[38]。
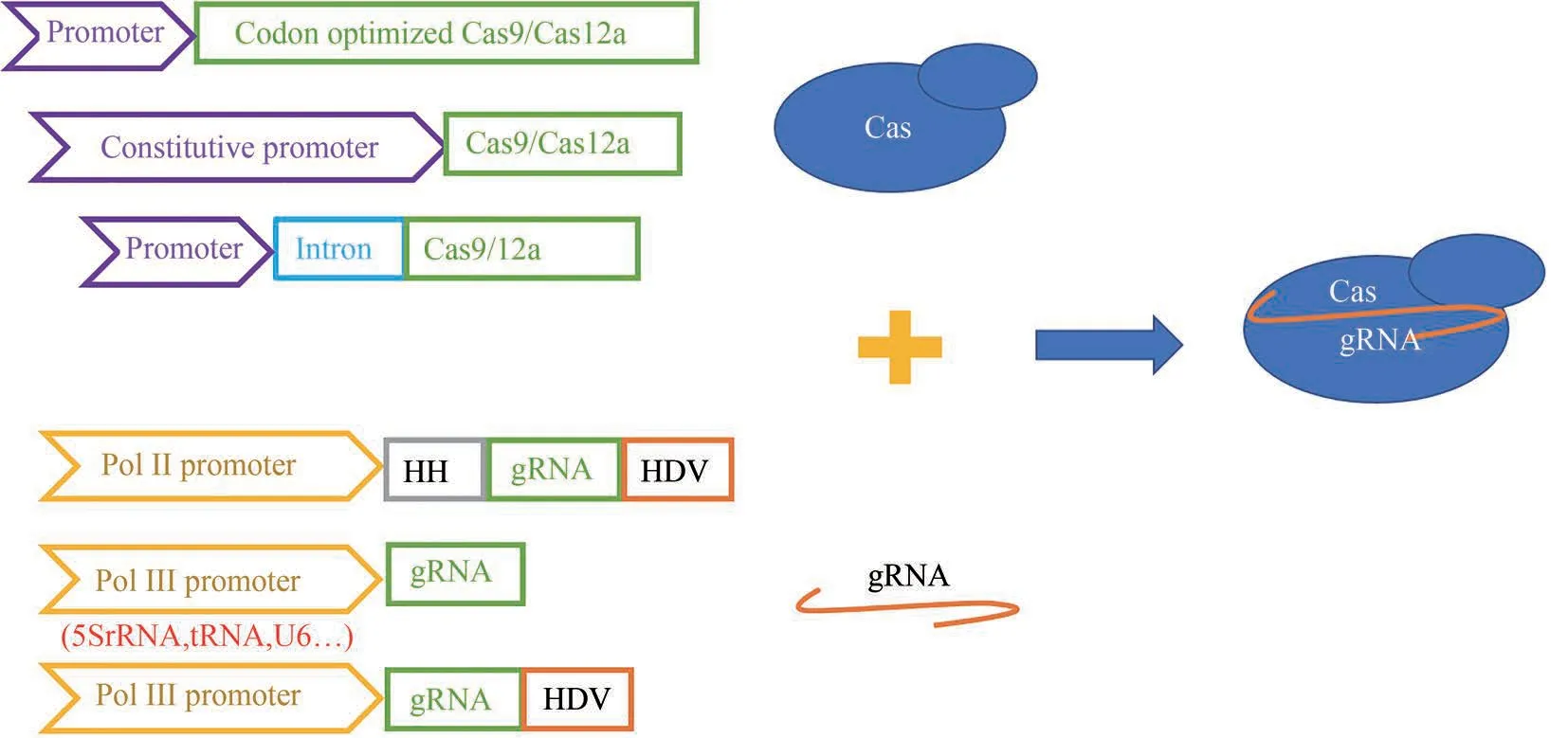
图2 CRISPR-Cas系统在丝状真菌中体内表达策略(针对Cas 蛋白的体内表达,有密码子优化,采用组成型启动子驱动蛋白表达和在Cas 基因5'端引入内含子等策略。针对gRNA 的体内表达,有用RNA 聚合酶II型启动子联合具有自我剪切功能的核酶表达gRNA、RNA 聚合酶III型启动子表达gRNA、RNA 聚合酶III型启动子联合具有自我剪切功能的核酶表达gRNA等策略)Fig.2 Strategies for in vivo expression of the CRISPR-Cas system in filamentous fungi(For in vivo expression of Cas protein,strategies include codon-optimization,adopting constitutive promoter for driving the expression of Cas protein,and incorporating intron sequence at the 5' end of cas gene. For in vivo expression of gRNA, strategies include adopting polymerase II promoter together with the self-cleaving ribozymes for gRNA expression,adopting polymerase III promoter for gRNA expression,and adopting polymerase III promoter together with the self-cleaving ribozyme for gRNA expression)
1.3 同源臂的设计
当CRISPR-Cas 系统产生DNA 双链缺口(double-strand break,DSB)时,宿主会通过非同源末端连接(none homologous end joining,NHEJ)或同源介导的修复(homology-directed repair, HDR)修补缺口(图3)。尽管经NHEJ 能在切割位点处缺失或插入片段可导致靶基因失活,但这样的编辑方式具有随机性,且不能引入研究者想要的变化。相比之下,HDR 则更为重要,因为在CRISPR-Cas 产生DSB 的同时引入HDR 供体修补缺口,宿主则有可能按照研究者的设计精准编辑靶位点,为研究基因功能、定点突变等奠定基础。关于同源重组供体的设计,在丝状真菌中以线性双链DNA 为主,通常在中心位置引入突变,两侧的同源臂紧邻基因组DSBs,长度为1~2kb 左右[15,49-52]。也有不少研究发现不超过40 bp 的短同源臂就能协助CRISPR-Cas 实现多种曲霉属丝状真菌的精准基因编辑[53-54]。Dong等[54]还发现在黑曲霉中,同源臂距离双链缺口越近,基因编辑效率越高。具体而言,他们设计了三段线性的双链DNA供体,供体中间是抗性筛选标记基因hygB,两侧同源臂长度均为39 bp,距离切口分别为0 bp、1 kb和5 kb,经CRISPR-Cas 介导的hygB 的整合效率分别是80%、50%和10%。除了线性同源重组供体外,Katayama 等[29]利用环状DNA 供体在米曲霉(A. oryzae)中成功实现基因编辑(图3)。此外,在里氏木霉(Trichoderma reesei)中,环状DNA供体也可介导高效的基因编辑[14]。有研究者发现,和线性DNA供体相比,环状DNA供体介导的靶基因编辑效率更高。他们推测原因是完整的环状DNA 由于缺少DSB,不大会通过在丝状真菌中占有主导地位的NHEJ修复途径整合到染色体上,因此,降低了筛选的假阳性率[39]。
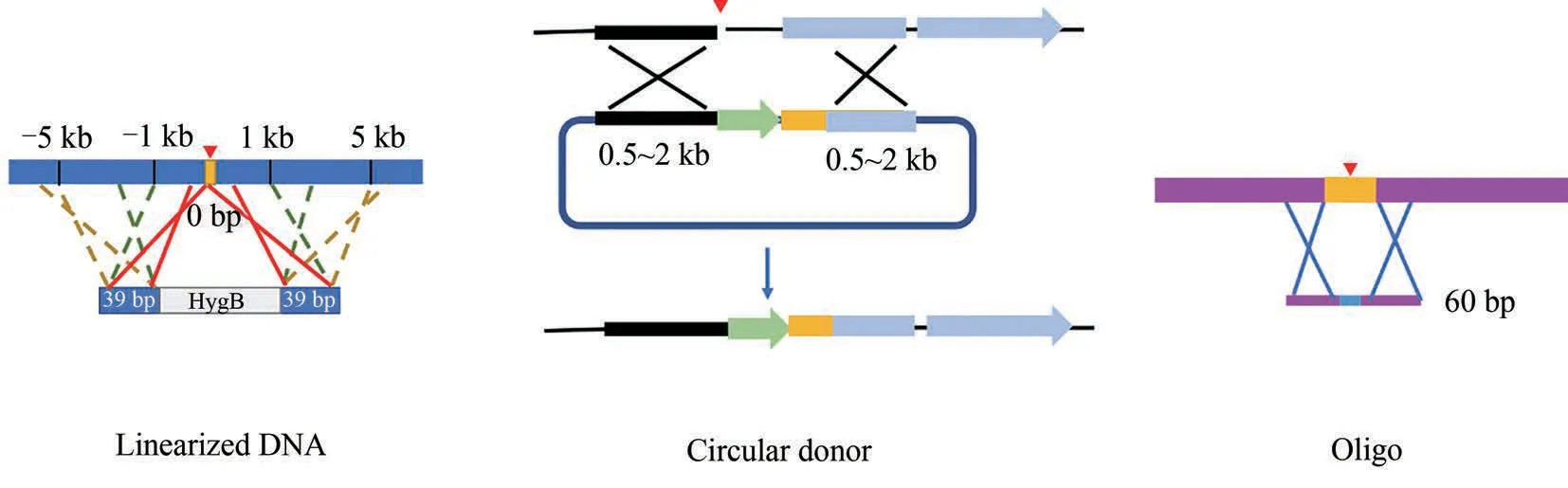
图3 同源重组供体的设计(线性双链DNA 供体,其两侧同源臂长39 bp, 距离CRISPR-Cas 系统产生的DNA 双链缺口5 kb 以内;环状DNA 供体,两侧同源臂长约0.5~2 kb,中间往往包含筛选标记;单链寡核苷酸,供体中突变位点和PAM之间距离不超过7 bp)Fig.3 Design of homologous recombination donor(For linearized DNA,it is flanked by 39 bp HR arms,which are within 5 kb from DSB as generated by CRIPSR-Cas.For circular DNA,it is flanked by HR arms with the size from 0.5 to 2 kb, and a selective marker is usually contained in the middle. For oligo, the intended mutation and PAM sequence are interspaced by no more than 7 bp)
近年来,单链寡核苷酸也被设计用于CRISPRCas 编辑丝状真菌基因时的同源重组供体。Nødvig等[55]设计了90 bp 的寡核苷酸链GE-Oligo1 和GEOligo2,分别与CRISPR-Cas 系统靶向的yA 基因的反义链和正义链互补,在供体中心引入了AAC 至TAA 的无义突变。无论转化GE-Oligo1 还是GEOligo2,大于90%的转化子都表现出yA 基因中断的表型。随机挑取基因中断菌株测序发现,当转化GE-Oligo1时,12个随机挑取的基因中断株中有10 株发生了和设计一致的变化,另外两株除了包含设计的变化以外,一株在TAA 下游3 个碱基处发生了单碱基缺失,另一株在TAA 下游插入了69个碱基的重复序列;当转化GE-Oligo2时,所有随机挑选基因中断菌株都发生了和设计一致的变化,且没有其他变化发生[55]。最近的一项研究表明,仅60 bp 长度的单链寡核苷酸也可以协助CRISPR-Cas 系统精准改变黑曲霉(A.niger)的靶序列,但需要精巧的设计。Kun 等[56]首先利用设计了90 bp 的单链寡核苷酸修复CRISPR-Cas 系统切割黑曲霉转录因子gaaR 的缺口,该供体包含了两处点突变:一处是CRISPR-Cas 系统识别的PAM;另一处是距离PAM位点52 bp的突变。遗憾的是用该供体转化获得的转化子只在PAM 的位点发生了突变,离它较远的突变并没有发生,保持同样的设计,将寡核苷酸链长度增加至200 bp 仍然不能获得预期的变化,这说明这些寡核苷酸修复模板并没有完全插入到双链缺口中。鉴于此,Kun 等重新设计了长度为60 bp 的寡核苷酸供体,将PAM 和预期突变位点的距离缩短至7 个碱基。在挑取的5 个转化子中,4 个发生了预期变化,仅有1 个转化子只发生了PAM 位点的突变。他们还设计了另一条60 bp 的寡核苷酸修补模板,除了包括上述2 个突变外,在距离PAM 15 bp 范围以内还增加了5 处点突变。在随机挑取的5 个转化子中,同样有4 个都发生了预期的7 处改变。综上,研究者推测距离DSBs 5'端不长于12个碱基的序列,更倾向于作为同源重组的修补模板[56]。
1.4 宿主的改造
在CRISPR-Cas 编辑丝状真菌的研究中,针对宿主改造的研究尚不多见。如前所述,NHEJ 和HDR 是宿主修复DSB 的两种形式,抑制或敲除NHEJ是大幅提高宿主HDR的有效策略,也是目前发现通过改造宿主细胞来提高CRISPR-Cas 精准编辑丝状真菌基因效率的唯一策略。研究者们通常使用NHEJ 途径失活的丝状真菌突变体为出发菌株,构建CRISPR-Cas 系统[56-57]。但是,也有研究发现在NHEJ 没被抑制的丝状菌株中,CRISPRCas 系统介导的微同源重组同样能高效地发生[53](表1),这提示我们可能存在不与NHEJ 竞争的多种同源重组修复途径。如何改造宿主对DSBs 进行高效的修复,可能还需依赖CRISPR-Cas 等工具首先解析丝状真菌中的相关修复途径,才可能提出行之有效的策略。
2 CRISPR-Cas系统编辑丝状真菌的挑战及可能的解决方法
CRISPR-Cas 系统改变了外源片段在丝状真菌中的整合方式,不仅大幅提高多种模式菌株如构巢曲霉(A. nidulans)、里氏木霉(T. reesei)等基因编辑的效率,甚至实现了传统方法无法编辑的、遗传操作困难的非模式菌株如灵芝(G. lucidum)等的精准基因编辑[9,15]。尽管如此,CRISPR-Cas系统并没有改变外源片段转化丝状真菌的效率,此外,编辑效率低也是CRISPR-Cas 系统在非模式丝状真菌中应用的另一大挑战(表1),本小节将针对这些挑战分析可能的原因并提出对应的解决办法。
2.1 转化效率低
丝状真菌的细胞壁是吸收外源DNA 的障碍,人们往往以去掉细胞壁的原生质体作为受体接受外源片段,而原生质体的低再生率限制了转化效率[58]。尝试开发不依赖于原生质体的递送外源片段的方式可能是一种解决方法,这可以从其他难操作宿主的递送方式的有关研究中获得启发。在2015 年的一项研究中,研究者设计了菱形缩颈的芯片,当细胞通过比其直径小的缩颈时,经历快速的机械变形可形成短暂的膜破裂或孔洞,gRNA和Cas 蛋白在此时可以从周围的介质进入到细胞中。利用该方法,Han等[59]成功将CRISPR-Cas系统递送到了多种细胞中,包括难于转染的淋巴瘤细胞和胚胎干细胞。此外,向自然界学习病毒侵染丝状真菌的过程[60],也可开发高效的、不依赖于原生质体转化的外源片段递送方式,其关键在于系统鉴定病毒在丝状真菌体内的复制元件。退而求其次,即便经原生质体再生限制了转化效率,挖掘菌株特异的自主复制元件用于装载外源片段,也能极大地提高转化效率。在前文中提到,AMA1是曲霉属中唯一发现的自主复制元件,有研究表明它能把黑曲霉(A.niger)的转化效率提高10~100 倍[61], 也 可 在 产 黄 青 霉 菌(Penicillium chrysogenum)中实现高效的基因敲除[20]。
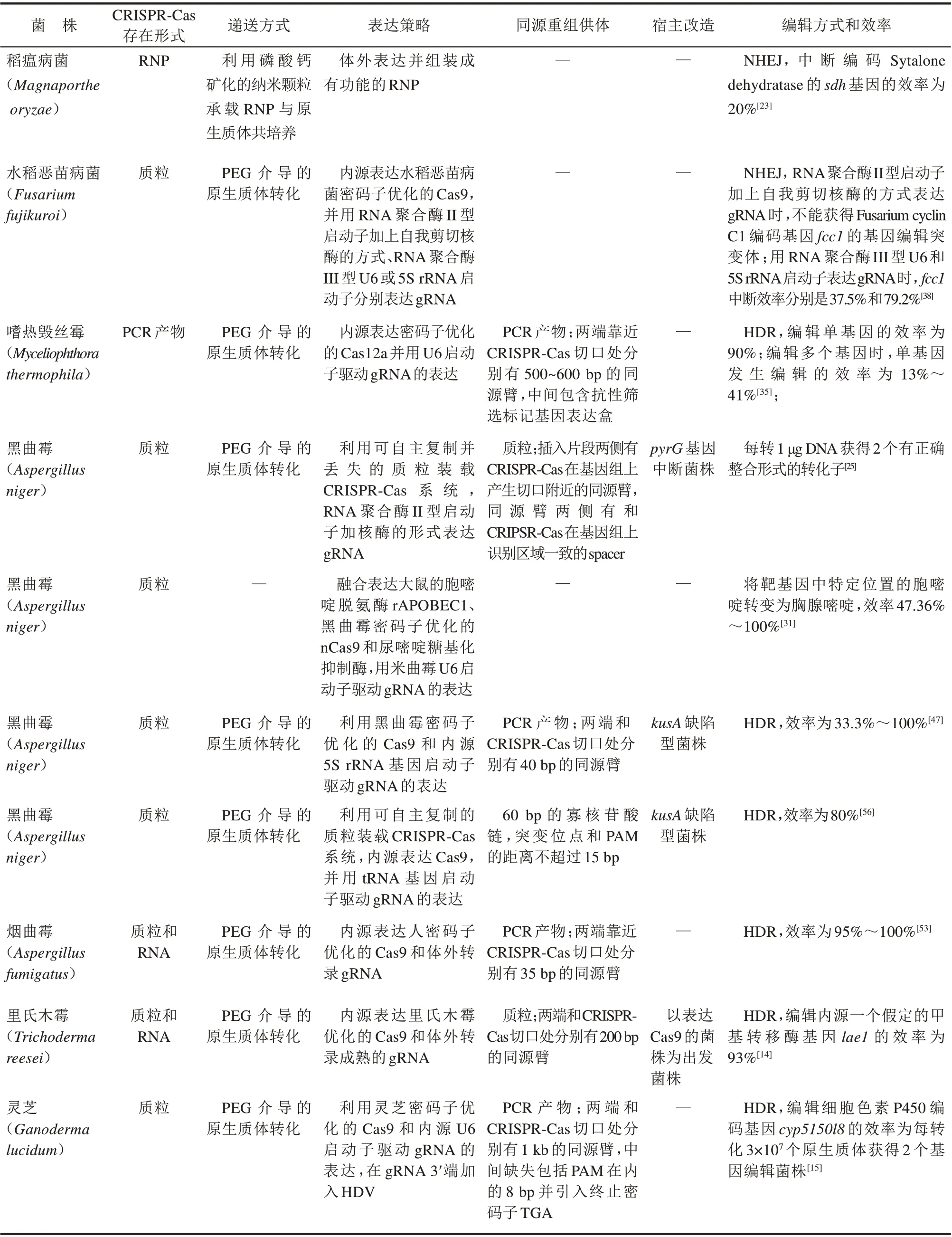
表1 CRISPR-Cas系统编辑丝状真菌的典型例子Tab.1 Examples of CRISPR-Cas system assisted gene editing in filamentous fungi.
2.2 编辑效率低
CRISPR-Cas 系统编辑一些丝状真菌的效率较低,可能有以下几方面原因:第一,CRISPR-Cas系统发挥功能的前提是Cas 蛋白和gRNA 同时进入同一个细胞核对DNA 进行切除,很多丝状真菌具有异核性,中断其中一个细胞核的基因并不意味着另一个细胞核的基因能被有效中断,这样的菌株往往因为仍然具有该基因的功能而不被筛选获得;第二,丝状真菌内源的防御机制阻碍CRISPRCas 系统进入细胞;第三,丝状真菌内源的防御机制包括重复序列诱发的点突变和RNA 沉默等[62],抑制了CRISPR-Cas系统的活力,导致即便CRIPSR-Cas系统同时进入所有细胞核,它们却不能有效行使靶向和/或切割功能,进而降低了编辑效率。
针对第一点,通过孢子分离等手段获得单核、单倍体的突变体,以它为出发菌株进行基因编辑,或许是一种解决办法。针对第二点和第三点,消除或缓解丝状真菌的多种内源防御是建立在对其充分认识和理解基础之上的。而正因为不清楚防御机制的关键遗传决定因子,导致某些丝状真菌的编辑效率无法得到显著提高。鉴于此,构建亲缘关系相近的模式丝状真菌的全基因组基因中断突变体库,利用报告系统考察突变体的表型,将有效地协助研究者解决上述问题。
3 总结与展望
丝状真菌成熟基因编辑平台的构建,对挖掘这类微生物在工业、农业、医药等多种行业的应用价值与潜力起关键作用。CRISPR-Cas 系统由于其组成和设计简单、特定靶向的优势,近年来在多种模式和/或非模式丝状真菌中得到越来越多的应用。基于CRISPR-Cas 系统开发的丝状真菌基因编辑形式包括特定位点的插入[24-25]、缺失[9]、碱基转换[31]和转录激活[32]。编辑的基因涵盖了易筛选的标记基因[14],非筛选标记的其他功能基因[15],甚至功能未知的基因[63],同时编辑的5个基因[33]。编辑的尺度可以是改变1 个碱基[55-56],也可以是缺失长达48 kb的基因簇[47]。在此基础上,通过中断宿主NHEJ 的关键基因和精妙的同源重组供体设计,使得CRISPR-Cas 系统能在特定位点引入变化,提高了编辑的精准性。
CRISPR-Cas 系统确实提高了不少丝状真菌的同源重组效率,在有些菌株中能达到100%的编辑效率,节省了筛选标记的使用,克服了这类微生物可使用筛选标记少的限制[35]。然而,在大多数非模式丝状真菌中,CRISPR-Cas 系统的基因编辑效率并不高[15,64],很可能是因为人们对这些难操作的宿主的代谢调控、防御机制认识有限,尚不清楚宿主特异性限制CRISPR-Cas 功能的决定因素。如2.2 中所讨论的,构建全基因组范围的基因中断突变体库将为解决这一基础科学问题奠定基础。关于构建全基因组范围的突变体库,我们目前已在一些模式丝状真菌如Aspergillus nidulans[65]、Neurospora crassa[66]等中看到希望。
致谢:感谢科技部“国家重点研发计划”(2018YFA09 00600)、国家自然科学基金(319713144和31600071)和上海市自然科学基金(17ZR1448900 和18ZR1420300)对肖晗博士的资助。感谢上海交通大学钟建江教授等的支持和有益讨论。
