Secologanol酰化产物的合成及其反应条件研究
2021-05-20杨宗斌郭海慧王福生
王 乐,杨宗斌,王 甜,郭海慧,王福生
(大理大学药学院,云南大理 671000)
Secologanol是裂环烯醚萜苷类成分,最早于1989年由法国学者从龙胆科龙胆属植物春龙胆(Gentiana verna)中发现〔1〕。在后续的研究中,有学者从龙胆科双蝴蝶属植物双蝴蝶(Tripterospermum chinense)〔2〕和刺莲花科岩荨麻属植物岩荨麻(Euc⁃nidebartonioides)〔3〕中也分离得到该化合物,但含量极低。本课题组前期对龙胆属植物微籽龙胆(Genti⁃ana delavayi)的研究中发现Secologanol为量丰化合物,进一步研究发现它能显著抑制人APP∕PS1 CHO细胞产生Aβ40寡聚体,具有潜在的研究开发价值〔4〕。由于环烯醚萜苷类成分的极性大,脂溶性和稳定性较差,而糖基修饰能提高糖苷类成分的化学稳定性〔5〕,加之,本课题组发现,环烯醚萜苷糖基不同位置酰化的天然产物具有不同的抗阿尔茨海默病(Alzheimer's disease,AD)活性〔6〕,为此,本实验拟对Secologanol进行结构修饰,合成其相应酰化产物,以期增强其衍生物的结构稳定性及抗AD活性,为进一步对它进行结构优化打下基础。
有学者对裂环马钱子苷(secoxyloganin)进行转化过程中,曾得到Secologanol的糖基四乙酰化产物2〔7〕,其方法是:先将裂环马钱子苷进行糖基乙酰化,再将7-位羧基还原得到化合物2,反应中7-位羧基还原为伯醇羟基较难,需要用氯甲酸乙酯活化才能反应。为此,本实验对Secologanol的乙酰化和苯甲酰化产物的合成条件进行了探讨,结果发现,通过控制反应条件,可以快速、高效得到化合物2及4个新的Secologanol酰化产物1、3、4和5。其合成路线、反应条件及结构见图1。
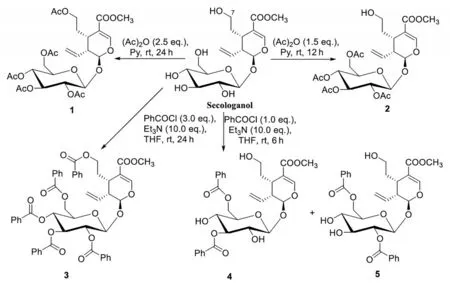
图1 Secologanol酰化产物的合成路线、反应条件及其结构
1 实验部分
1.1 仪器与试剂 仪器:磁力搅拌器(北京大龙兴创实验仪器股份公司);AvanceⅢ-400型核磁共振波谱仪(德国布鲁克公司);循环水式真空泵(巩义市予华仪器有限公司);旋转蒸发仪(上海亚荣生化仪器厂);分析天平(梅特勒-托利多仪器有限公司)。
试剂:Secologanol由本课题组自己制备;吡啶、乙酸酐、碳酸氢钠(NaHCO3)、苯甲酸、氯化亚砜(SOCl2)、二甲基甲酰胺(DMF)、四氢呋喃、三乙胺、乙酸乙酯、无水硫酸钠均购自国药集团化学试剂有限公司;氘代氯仿(CDCl3)购自北京百灵威科技有限公司。
1.2 实验步骤
1.2.1 化合物1和2的合成 称取化合物Secologanol 500 mg(1.28 mmol),装入25 mL圆底烧瓶中,加入4 mL吡啶充分溶解,再加入乙酸酐181µL(1.92 mmol),在氮气保护下室温反应约12 h。薄层色谱法(TLC)检测,待反应原料消失,发现有1个主要产物生成,加入饱和NaHCO3溶液淬灭反应,用乙酸乙酯萃取5次,合并有机相,无水硫酸钠干燥,减压浓缩,残余物经硅胶柱色谱(石油醚:乙酸乙酯=2:1)纯化得到化合物2(白色固体610.5 mg)和化合物1(白色固体65 mg)。收率按以下公式计算:收率=实际产量∕理论产量×100%。化合物1和2的收率分别为8.5%和85.3%。
同上,称取化合物Secologanol 50 mg(0.13 mmol),装入10 mL圆底烧瓶中,加入1 mL吡啶充分溶解,再加入乙酸酐31µL(0.33 mmol)。在氮气保护下室温反应约24 h。TLC检测,反应至仅有1个主要产物,经处理及柱层析后得到化合物1(白色固体67.8 mg,收率90.6%)。
为了验证化合物2是否能完全转化为化合物1,称取化合物2 50 mg(0.09 mmol)置于10 mL圆底烧瓶中,加入1 mL吡啶充分溶解,再滴加乙酸酐22µL(0.23 mmol),在氮气保护下室温反应约24 h,TLC检测,待原料消失,发现仅有1个主要产物。同上方法处理,经硅胶柱色谱(石油醚:乙酸乙酯=2:1)纯化得到化合物1(白色固体45.2 mg,收率83.7%),证明化合物2也可以完全转化为化合物1。
化合物1:白色固体;1H-NMR(400 MHz,CDCl3)δ:7.32(s,1H),5.57(dt,J=17.1,9.8 Hz,1H),5.31(d,J=1.4 Hz,1H),5.28~5.23(m,3H),5.18(dd,J=9.5,0.8 Hz,1H),5.09(td,J=9.7,0.9 Hz,1H),4.99(ddd,J=9.5,8.1,1.0 Hz,1H),4.85(d,J=8.1 Hz,1H),4.27(dd,J=12.4,4.5 Hz,1H),3.68(s,3H),3.28(dd,J=4.1,0.9 Hz,1H),2.70(dt,J=40.8,4.6 Hz,2H),2.08(s,3H),2.03(s,3H),2.00(s,3H),1.98(s,3H),1.90(s,3H);13C-NMR(100 MHz,CDCl3)δ:171.56,171.06,170.63,169.79,169.41,167.33,151.03,133.04,120.81,111.30,96.43,96.10,72.50,72.56,70.87,68.47,62.61,62.02,51.00,73.00,43.26,27.86,27.64,21.37,21.18,21.01,20.63。
化合物2:白色固体;1H-NMR(400 MHz,CDCl3)δ:7.51(d,J=2.4 Hz,1H),5.42(dt,J=17.0,9.8 Hz,1H),5.28(t,J=2.2 Hz,2H),5.18(d,J=9.5 Hz,1H),5.04(t,J=9.7 Hz,1H),4.95(t,J=8.8 Hz,1H),4.87(d,J=8.1 Hz,1H),4.40(dt,J=11.2,3.3 Hz,1H),4.09(dd,J=12.5,2.1 Hz,1H),3.72(s,3H),2.64(ddd,J=9.7,5.7,1.7 Hz,1H),2.05(s,3H),1.98(s,3H),1.96(s,3H),1.92(s,3H);13C-NMR(100 MHz,CDCl3)δ:170.20,170.16,170.01,169.80,166.23,150.03,132.04,120.01,110.30,96.33,95.80,72.30,72.16,70.67,67.47,63.61,63.12,50.00,53.00,42.06,27.56,27.24,21.27,21.11,20.61,20.53。1.2.2 化合物3、4和5的合成 苯甲酰氯的制备〔8〕:称取苯甲酸500 mg(4.10 mmol)装入25 mL圆底烧瓶中,氮气保护下滴加1滴干燥的DMF作为催化剂,随后在冰浴条件下缓慢地滴加SOCl2893µL(12.29 mmol),使其充分混溶。升至室温后反应1.5 h,再加热至80℃回流反应4 h。反应完成后,减压除去过量的SOCl2,得黄色油状物(558 mg,收率97.3%)。
化合物3、4和5的合成:称取Secologanol 100 mg(0.26 mmol)装入10 mL圆底烧瓶中,在氮气的保护下,用2 mL四氢呋喃溶解,然后滴加三乙胺358µL(2.60 mmol),在冰浴条件下缓慢滴加苯甲酰氯36 mg(0.26 mmol),室温反应约6 h后原料消失,TLC检测发现主要产物为衍生物4和5。延长反应时间至12 h,发现有新衍生物3生成,衍生物4和5的量减少。加入饱和NaHCO3溶液淬灭反应,用乙酸乙酯萃取5次,合并有机相,无水硫酸钠干燥,减压浓缩,残余物经硅胶柱层析(石油醚:乙酸乙酯=2:1)纯化得到化合物3(白色固体48.9 mg,收率21.0%)、化合物4(白色固体39.7 mg,收率26.1%)、化合物5(白色固体43.7 mg,收率29.0%)。
为了验证Secologanol是否能完全转化为化合物3,称取Secologanol 50 mg(0.13 mmol)装入25 mL圆底烧瓶中,在氮气的保护下,用1 mL四氢呋喃溶解,滴加三乙胺179µL(1.30 mmol)。滴加完毕后,冰浴条件下缓慢滴加苯甲酰氯55 mg(0.39 mmol),室温反应约24 h。待原料消失,发现仅有1个主要产物生成,残余物经硅胶柱层析(石油醚:乙酸乙酯=2:1)纯化得到化合物3(白色固体95.1 mg,收率81.9%)。证明Secologanol可以完全转化为化合物3。
化合物3:白色固体;1H-NMR(400 MHz,CDCl3)δ:8.07(d,J=1.2 Hz,2H),8.05(d,J=1.6 Hz,2H),8.03(d,J=1.2 Hz,1H),8.01(d,J=1.5 Hz,2H),7.90(ddd,J=8.4,4.1,1.4 Hz,7H),7.84(d,J=1.3 Hz,2H),7.82(d,J=1.5 Hz,2H),7.41(s,1H),7.13(d,J=1.5 Hz,2H),5.91(d,J=9.6 Hz,1H),5.71(t,J=9.7 Hz,2H),5.35(d,J=4.5 Hz,2H),5.32(d,J=1.4 Hz,1H),4.66(d,J=3.2 Hz,1H),4.63(d,J=3.1 Hz,1H),4.53(d,J=5.1 Hz,1H),4.50(d,J=5.0 Hz,1H),3.34(s,3H),2.93~2.82(m,1H),2.78~2.62(m,1H);13C-NMR(100 MHz,CDCl3)δ:166.57,166.41,166.11,165.74,165.15,164.71,150.77,133.53,133.32,133.23,133.10,132.87,132.78,130.34,130.12,129.84,129.78,129.77,129.74,129.69,129.47,129.12,128.99,128.70,128.65,128.45,128.42,128.34,128.31,126.28,120.30,110.60,96.68,96.57,72.63,72.53,71.15,69.50,62.94,62.83,51.02,43.03,31.95,29.69。
化合物4:白色固体;1H-NMR(400 MHz,CDCl3)δ:8.03(dt,J=7.0,1.4 Hz,1H),8.00(d,J=1.2 Hz,1H),7.99~7.97(m,1H),7.95(d,J=1.5 Hz,1H),7.91(ddd,J=8.4,4.0,2.5 Hz,1H),7.49(dt,J=7.5,1.9 Hz,1H),7.47~7.44(m,1H),7.40~7.32(m,2H),5.64~5.59(m,1H),5.43(d,J=6.6 Hz,1H),5.01(d,J=7.8 Hz,1H),3.92(s,1H),3.65(s,3H),3.31~3.24(m,1H),3.15(s,1H);13C-NMR(100 MHz,CDCl3)δ:167.82,165.85,165.79,150.12,133.69,133.04,133.01,130.14,130.11,129.89,129.87,129.86,129.81,128.57,128.55,128.52,128.46,120.46,111.26,109.04,79.58,74.38,73.45,71.24,68.86,44.87,33.52,30.65,29.01,28.34。
化合物5:白色固体;1H-NMR(400 MHz,CDCl3)δ:8.04(dd,J=8.3,1.4 Hz,1H),7.91(ddd,J=8.5,3.5,1.4 Hz,2H),7.86(dd,J=8.4,1.4 Hz,1H),7.51(s,1H),7.46~7.40(m,2H),7.39~7.27(m,2H),5.96(t,J=9.8 Hz,1H),5.74(t,J=9.7 Hz,1H),5.56(dd,J=10.0,8.1 Hz,1H),5.40(d,J=1.3 Hz,1H),5.29~5.19(m,2H),4.66(dd,J=12.3,3.0 Hz,1H),4.53(d,J=4.8 Hz,1H),4.26~4.18(m,1H),4.10(dd,J=12.9,7.0 Hz,1H),3.48(s,3H),2.67~2.58(m,1H);13C-NMR(100 MHz,CDCl3)δ:167.19,165.85,165.85,150.12,133.69,133.04,133.04,130.14,130.14,129.89,129.89,129.86,129.86,128.57,128.55,128.52,128.52,120.46,111.30,108.70,80.20,73.36,72.19,69.20,61.10,52.29,44.90,33.52,30.68,28.60。
2 结果与讨论
在对Secologanol进行酰化反应过程中,发现酰化剂的量及反应时间均对反应产物产生较大影响。在乙酰化反应中,当酰化剂为1.5当量,反应时间为12 h时,化合物2为主产物,收率为85.3%,而化合物1的收率仅为8.5%,说明Secologanol糖基上羟基相较7-位羟基更容易被酰化;当酰化剂为2.5当量,反应时间为24 h时,仅得到全乙酰化产物1且收率为90.6%。该结果说明,通过控制反应条件,可快速、高效得到化合物1和2。
在进行苯甲酰化过程中,当酰化剂为1.0当量,反应时间为6 h时,化合物4和5为主要产物;增加反应时间至12 h,有新衍生物3生成,化合物3、4和5的收率分别为21.0%、26.1%和29.0%。当苯甲酰氯的量为3.0当量,反应时间为24 h,反应仅得到全苯甲酰化产物3,收率为81.9%。该结果说明,通过控制反应条件,也可快速、高效得到化合物3、4和5。
在进行酰化反应时,需严格控制在无水条件下进行。全乙酰化产物1和四乙酰化产物2的高收率制备,为Secologanol的进一步结构修饰打下良好基础;其新苯甲酰化产物3、4和5的制备,为其活性研究提供了新的衍生物。Secologanol不同酰化产物1~5的生物活性有待进一步研究。
