氯胺酮、羟亚胺及邻氯苯基环戊酮合成工艺的判别研究
2021-05-17樊颖锋吴健美花镇东王优美
樊颖锋,吴健美,花镇东,王优美
(公安部禁毒情报技术中心 公安部毒品监测管控与禁毒关键技术重点实验室 北京 100193)
氯胺酮是苯环己哌啶的衍生物,为非巴比妥类静脉麻醉剂,属于N-甲基-D-天门冬氨酸非竞争性的受体拮抗剂[1],具有兴奋中枢神经作用,能产生分离性幻觉,在我国及东南亚国家滥用非常广泛。2018年,氯胺酮已成为我国仅次于甲基苯丙胺的第二大流行滥用的合成毒品[2]。目前,国际禁毒公约尚未将氯胺酮列入管制,我国已将氯胺酮列入《麻醉药品和精神药品品种目录》作为一类精神药品管制,并将氯胺酮的化学前体——邻氯苯基环戊酮和羟亚胺列入《易制毒化学品的分类和品种目录》第一类。
毒品特征分析是采用高精密仪器分析毒品中的特征杂质,并对分析数据进行统计计算的一类法庭科学分析方法。近年来,各国主要研究的毒品特征分析方法包括采用气相色谱-质谱(GC-MS)、液相色谱-质谱(LC-MS)等仪器分析海洛因[3-4]、甲基苯丙胺[5-6]和可卡因[7]等常见毒品的有机杂质,采用电感耦合等离子体-质谱(ICP-MS)分析海洛因的无机杂质[8],采用同位素质谱(IR-MS)分析海洛因和甲基苯丙胺的碳、氮等同位素的比例[9-10],以实现毒品来源地、毒品合成原料和合成工艺等的推断。毒品特征分析可为禁毒执法部门监测毒品市场、追溯毒品来源、推断毒品贩运路线及毒品案件关联性判别提供科学依据。
由于氯胺酮在欧美等国家滥用不广泛,管制级别低,执法打击力度小,因此有关氯胺酮及其化学前体特征分析的文献报道也极少[11],本实验室采用LC-MS检测筛查出氯胺酮17种未知特征杂质,并通过对杂质的半定量结果统计学分析,判别样品的案件关联性[12]。甲基苯丙胺通常由麻黄碱/伪麻黄碱和1-苯基-2-丙酮这两种合成前体,可通过催化加氢法、碘红磷法和还原胺法等多种工艺合成,判别合成前体和合成路线是甲基苯丙胺特征分析的重要研究内容[6]。氯胺酮的基本合成路线通常包含三步:第一步合成邻氯苯基环戊酮(邻酮),第二步将邻氯苯基环戊酮溴代后与甲基胺反应合成羟亚胺,第三步由羟亚胺加热使分子结构重排后得到氯胺酮。其中,第二步和第三步合成通常采用单一的合成工艺;第一步邻氯苯基环戊酮的合成通常有两种合成工艺,一种是邻氯苯甲腈与卤代环戊烷的格氏试剂反应,另一种是邻氯苯甲酰氯与环戊烯的傅克酰基化反应[13]。非法制毒工厂生产氯胺酮及其化学前体的主要路线见图1。
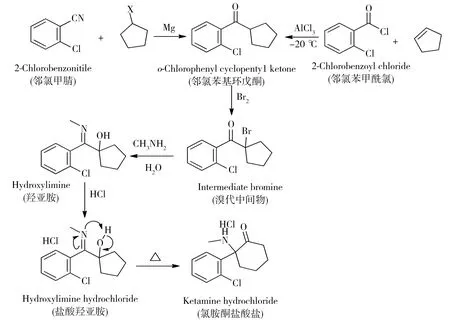
图1 非法制毒工厂氯胺酮及其化学前体合成路线Fig.1 Synthetic pathway of ketamine and its precursors in illicit lab
本文通过模拟非法工厂合成邻酮等方式,筛选邻氯苯甲腈与卤代环戊烷的格氏试剂合成邻酮时产生的特征杂质,并通过检测缴获邻酮、羟亚胺和氯胺酮样品中是否含有该特征杂质,判别其合成工艺,从而实现对非法制造氯胺酮、羟亚胺和邻氯苯基环戊酮合成工艺的监测。
1 实验部分
1.1 试剂与样品
氯代环戊烷和环戊酮(东京化成工业株式会社,日本),邻氯苯甲腈(北京百灵威科技有限公司),盐酸、氢氧化钠、三乙胺和四氢呋喃(北京化工厂),镁粉(山东西亚化学试剂公司),碘(成都化学试剂厂),甲醇、正己烷(Fisher Scientific公司,美国),所有试剂均为分析纯。8份邻氯苯基环戊酮样品和13份羟亚胺(盐酸盐)样品取自2014~2018年国内不同非法加工厂缴获样品,105份氯胺酮样品取自2016~2018年全国对应案件氯胺酮缴获量大于1 kg的样品。
1.2 邻氯苯基环戊酮合成实验
采用邻氯苯基环戊酮制造工厂常用格氏试剂方法的进行合成(合成路线见图2)。第一步:在三口烧瓶中加入1 mL氯代环戊烷、10 mL三乙胺、0.1 g碘、7 g镁粉和10 mL四氢呋喃,搅拌加热至70 ℃,引发反应;第二步:升温至80 ℃,加入15 mL三乙胺和30 mL四氢呋喃,缓慢滴加29 mL氯代环戊烷,回流反应3 h,得到格氏试剂;第三步:滴加26 g邻氯苯甲腈和80 mL四氢呋喃混合溶液,保持80 ℃,回流反应6 h;第四步:降至室温后加入200 mL水,再滴加10 mL浓盐酸,保持50 ℃,反应5 h;第五步:降至室温,取上层四氢呋喃溶液,以80 ℃旋蒸除去四氢呋喃,再升温至165 ℃除去邻氯苯基环戊酮,得到合成邻氯苯基环戊酮样品23.4 g,产率约52%。
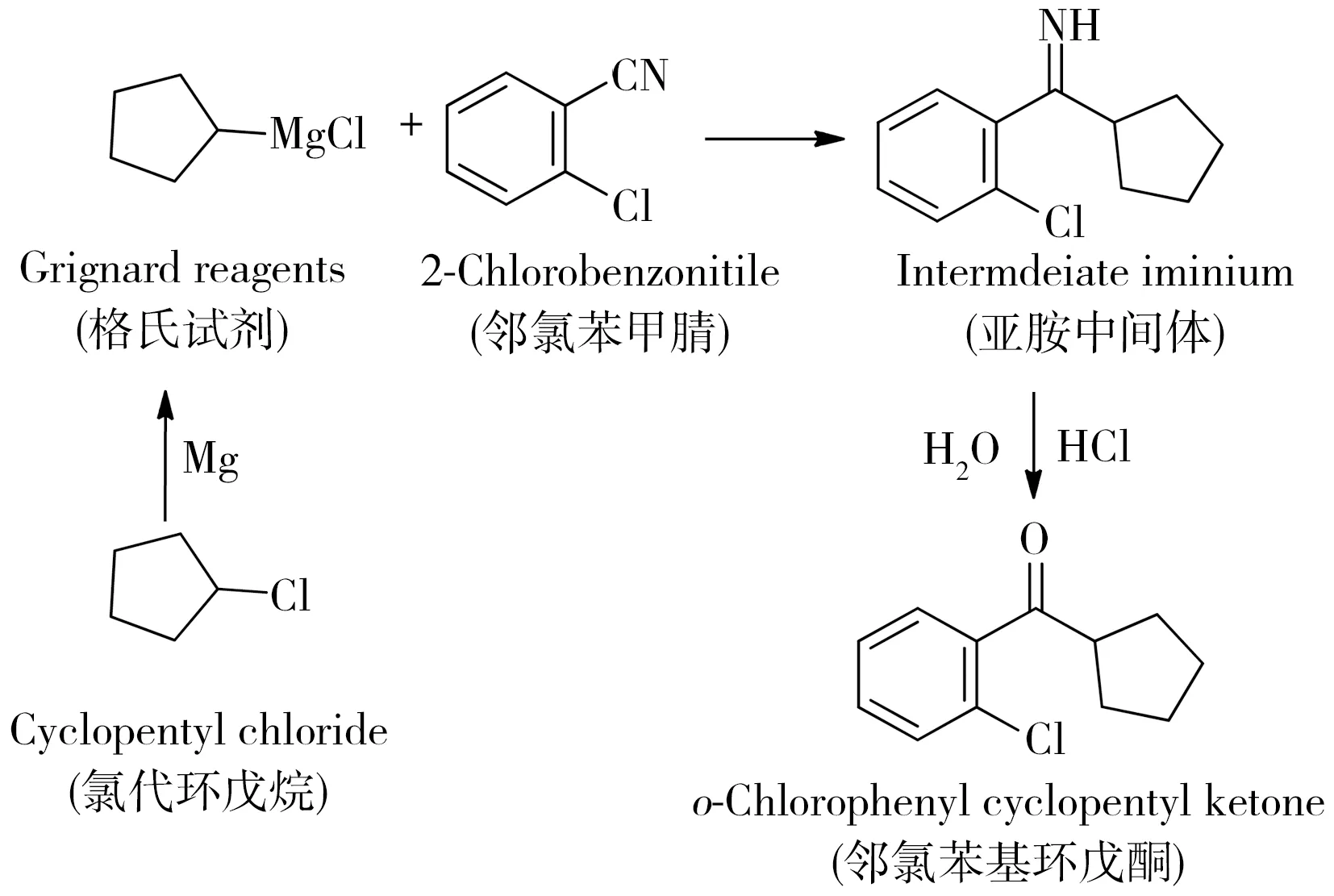
图2 邻氯苯基环戊酮的合成路线Fig.2 Synthesis of o-chlorophenyl cyclopentyl ketone by Grignard reagents routine in this study
1.3 邻氯苯基环戊酮与羟亚胺样品特征杂质分析
1.3.1 仪器条件QP2000 Ultra气相色谱-质谱联用仪分析(Shimadzu公司,日本),色谱柱Rtx-5ms(30 m×0.25 mm,0.25 μm);氦气流速1.0 mL/min,进样口温度280 ℃,进样体积1 μL;升温程序:起始温度60 ℃,保持2 min,然后以10 ℃/min升至280 ℃,保持5 min;分流进样,邻氯苯基环戊酮样品分流比100∶1,羟亚胺样品分流比10∶1,溶剂延迟2.1 min。质谱离子源为EI源,电离能量70 eV,离子源温度230 ℃,接口温度250 ℃,全扫描模式,扫描范围35~400 amu。为避免质谱检测器饱和,在邻氯苯基环戊酮和羟亚胺等主要化合物出峰时关闭质谱分析器。
1.3.2 邻氯苯基环戊酮样品前处理将邻氯苯基环戊酮样品混合均匀,吸取100 μL于具塞离心管中,加入10 mL甲醇,超声溶解,涡旋混匀后过0.45 μm有机系滤膜,待分析。
1.3.3 羟亚胺样品前处理将羟亚胺样品用研钵研磨均匀,称取100 mg于具塞离心管中,加入4 mL Tris溶液(pH 8.1)溶解,再加入200 μL正己烷,振荡、离心,取约100 μL上层有机溶液于进样瓶内插管中待用。
1.4 氯胺酮样品特征杂质分析
1.4.1 仪器条件HSS 86.50顶空进样器(DANI,意大利)和QP2000 Ultra气相色谱-质谱联用仪(Shimadzu公司,日本)分析。顶空进样器条件:辅助气压0.8×105Pa样品平衡温度(OVEN)、进样系统温度(MAIN)和传输管温度(TUBE)均为190 ℃,向顶空瓶加压时间、充满样品环时间和样品进入GC-MS时间均为10 s,样品加热平衡时间20 min,高速样品摇晃模式。气相色谱-质谱条件:色谱柱VF-624ms(60 m×0.25 mm,0.14 μm);氦气流速2.0 mL/min,进样口温度240 ℃,不分流进样;升温程序:起始温度80 ℃,保持2 min,然后以15 ℃/min升至250 ℃,保持10 min。质谱离子源为EI源,电离能量70 eV,离子源温度230 ℃,接口温度250 ℃,选择离子扫描模式(选择的特征离子见表1)。
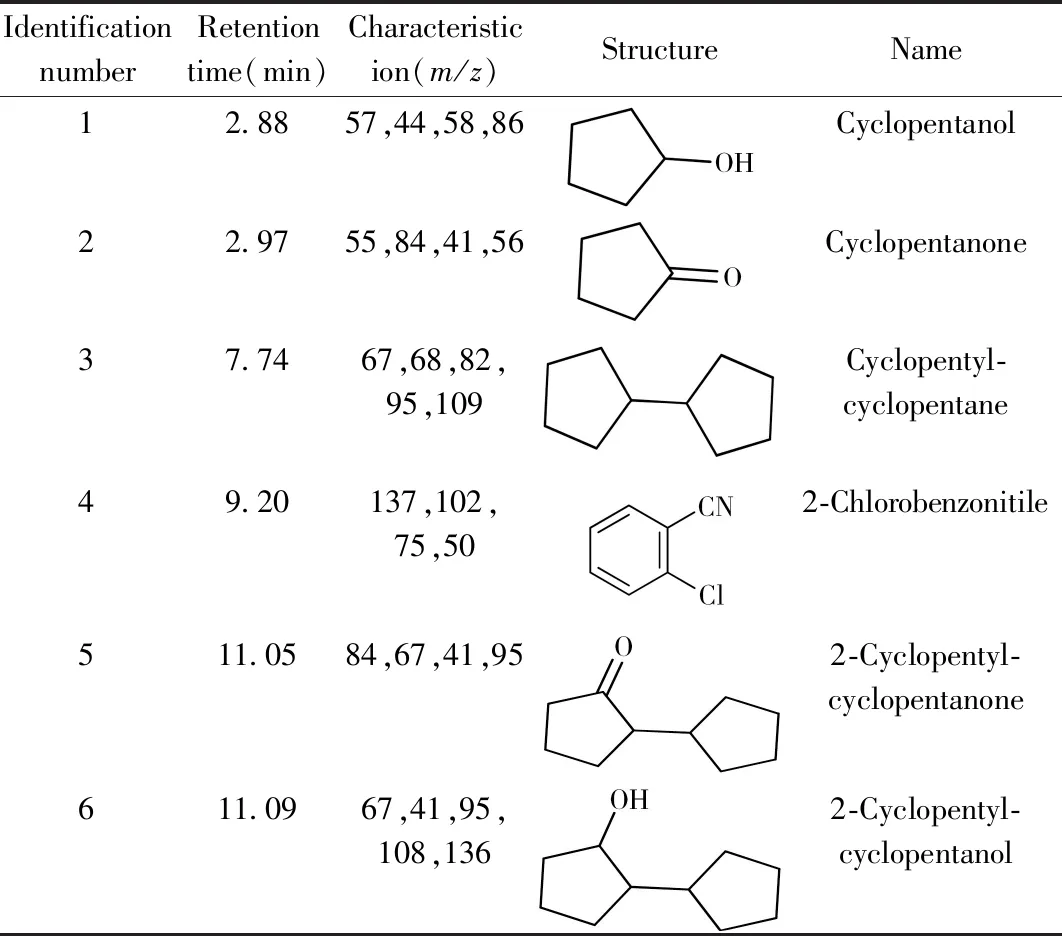
表1 格氏试剂路线获得的邻氯苯基环戊酮样品中的特征杂质Table 1 Profiling impurities found in o-chlorophenyl cyclopentyl ketone sample from Grignard reagents routine
1.4.2 氯胺酮样品前处理将氯胺酮样品用研钵研磨均匀,称取100 mg于顶空进样瓶中,密封,待分析。
2 结果与讨论
2.1 邻氯苯基环戊酮样品的特征杂质
缴获邻氯苯基环戊酮样品共8份,取自近年来国内8起缴获量较大的邻氯苯基环戊酮非法制造、贩卖案件,具有较强的代表性。8份缴获样品的TIC图中有机杂质峰均超过100个,其中信号较强的杂质有邻氯苯甲腈、邻氯苯甲醛、环戊酮和环戊基环戊烷等(图3和表1)。邻氯苯甲腈为格氏试剂法合成邻氯苯基环戊酮的主要原料(图1)。根据He等[14]报道,在有氯环境中格氏试剂连接卤素的位置可生成羟基或羰基,因此邻氯苯基环戊酮样品中的环戊酮和环戊醇可能为卤代环戊烷格氏试剂在合成反应中的副产物(图4)。高温条件下,卤代环戊烷格氏试剂可发生偶联反应,生成环戊基环戊烷[15],反应路线见图5。此外,双卤代环戊烷格氏试剂可能相继发生偶联反应和氧化反应,生成2-环戊基环戊酮和2-环戊基环戊醇(图6)。邻氯苯甲醛等苯环类的杂质可能来源于邻氯苯甲腈,也可能来源于傅克酰基化反应的主要原料——邻氯苯甲酰氯,因此这些杂质不适合作为判别工艺的特征杂质。
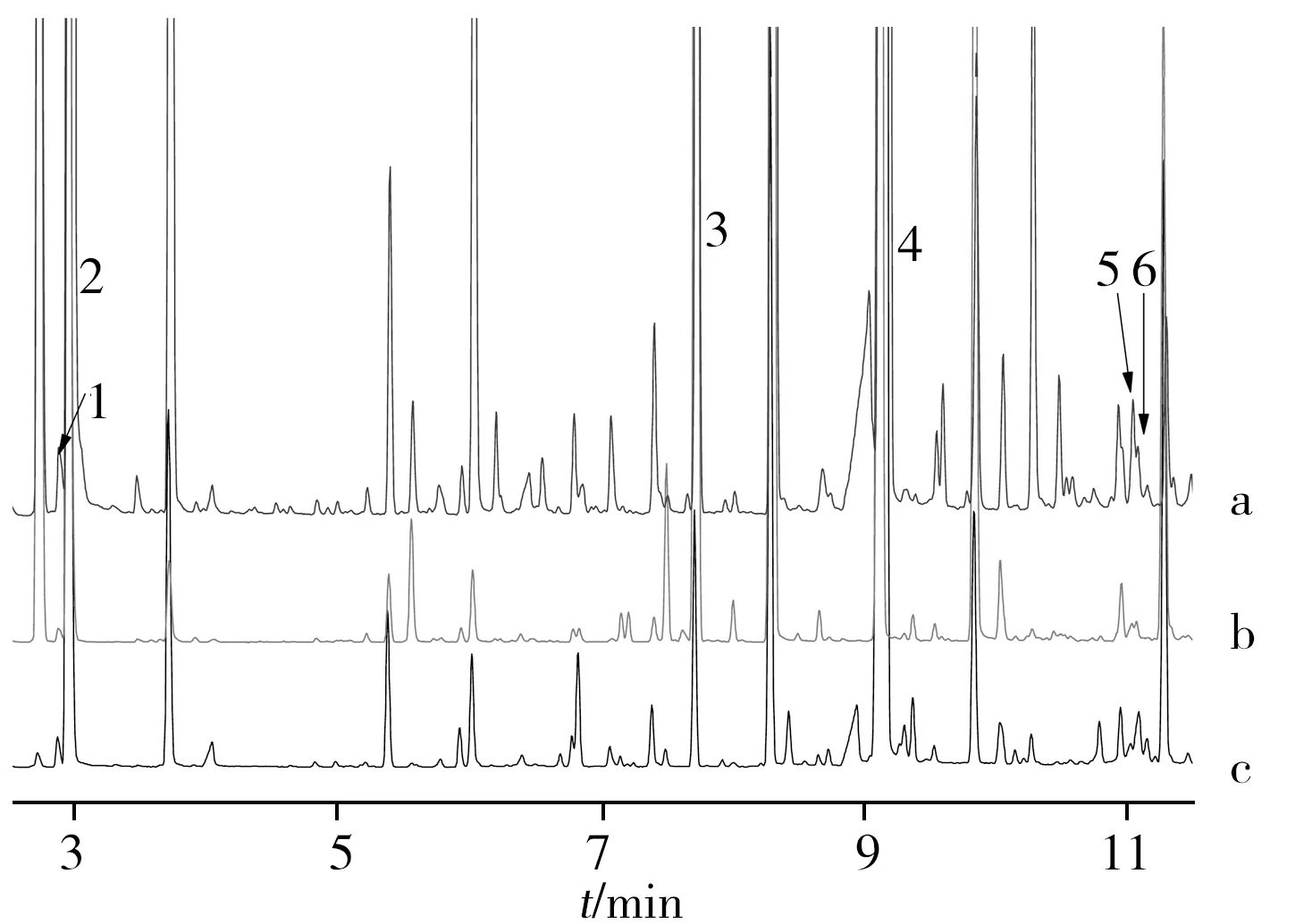
图3 缴获邻氯苯基环戊酮、合成邻氯苯基环戊酮和羟亚胺的TIC色谱图Fig.3 TIC chromatograms of seized o-chlorophenylcyclopentyl ketone,synthesized o-chlorophenyl cyclopentyl ketone and hdroxylimineThe identification number(1-6) of 6 peaks are the same as those in table 1;a:seize o-chlorophenylcyclopentyl ketone,b:synthesized o-chlorophenylcyclopentyl ketone,c:hdroxylimine

图4 环戊基格氏试剂与氧气的反应Fig.4 Reaction of Grignard Reagents and Oxygen
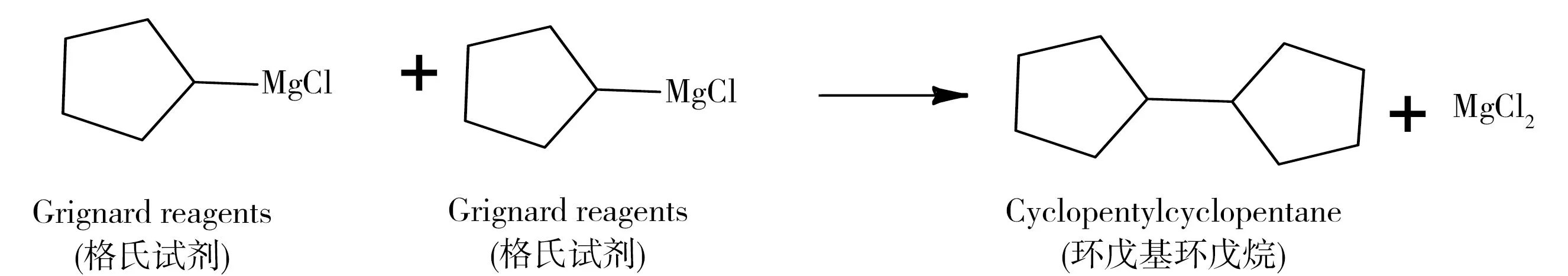
图5 环戊基格氏试剂的偶联反应Fig.5 Coupling reaction of Grignard Reagent

图6 环戊基格氏试剂的偶联和氧化反应Fig.6 Coupling and oxidation reaction of Grignard Reagents
为验证缴获的邻氯苯基环戊酮的合成工艺为格氏试剂法,本实验采用工厂常用的格氏试剂合成方法,以氯代环戊烷、镁和邻氯苯甲腈为主要原料合成了邻氯苯基环戊酮(合成路线见图2);其中,氯代环戊烷不含表1中6种杂质。比较缴获的邻氯苯基环戊酮和实验室合成的邻氯苯基环戊酮的TIC色谱图(图3a和b)发现,实验室合成的邻氯苯基环戊酮样品中杂质峰数量相对较少,信号相对较弱,检出环戊酮、环戊醇、环戊基环戊烷和邻氯苯甲腈,但未检出2-环戊基环戊酮和2-环戊基环戊醇,可能是由于使用的氯代环戊烷含1,2-邻氯环戊烷含量极少,使得产生的双卤代环戊烷格氏试剂极少。
2.2 羟亚胺样品的特征杂质
由于结晶过程中造成了大量损失,羟亚胺盐酸盐中的特征杂质含量很低,故采取甲基苯丙胺特征分析中常用的液-液萃取富集特征杂质[16]。结果显示,13份羟亚胺样品均检出环戊酮、环戊醇、环戊基环戊烷和邻氯苯甲腈4种特质杂质,其中羟亚胺的TIC色谱图见图3c。由此推测,13份羟亚胺样品使用的化学前体为邻氯苯基环戊酮,其合成工艺为格氏试剂与邻氯苯甲腈反应的方法。
2.3 氯胺酮样品的特征杂质
氯胺酮样品中,邻氯苯基环戊酮中残留的特征杂质极少,采用液-液萃取获得的萃取溶液仅少部分能进入GC-MS中分析,不足以检出特征杂质。因此,采用高温顶空进样的方法,在190 ℃加热和高速样品摇晃模式下使氯胺酮晶体中包裹的特征杂质挥发,并加压使大部分挥发性气体进入GC-MS中分析;同时在质谱分析器中采用SIM扫描模式检测特征杂质的特征碎片离子,得到的氯胺酮的TIC色谱图见图7,由图可见,大部分氯胺酮样品中检出环戊酮和邻氯苯甲腈,但未检出环戊醇和环戊基环戊烷,这可能因为环戊醇在邻氯苯基环戊酮样品中含量较低,而环戊基环戊烷极性弱,在羟亚胺和氯胺酮成盐及结晶过程中无法残留。最终在105个氯胺酮样品中全部检出环戊酮,102个样品中检出环戊酮和邻氯苯甲腈。由此推测,102个缴获氯胺酮样品使用的化学前体为邻氯苯基环戊酮,其合成工艺为格氏试剂与邻氯苯甲腈反应的方法。
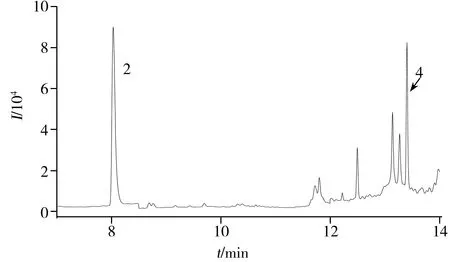
图7 顶空进样获得的氯胺酮样品TIC色谱Fig.7 TIC chromatogram of Ketamine by headspace injection the identification number(2,4) of 2 peaks are the same as those in table 1
3 结 论
本文采用多种仪器和样品前处理方法,并通过合成实验验证,分析并确证了邻氯苯基环戊酮、羟亚胺和氯胺酮样品中的特征杂质。结果表明,全部邻氯苯基环戊酮和羟亚胺样品,以及绝大多数氯胺酮样品均含有邻氯苯甲腈与格氏试剂反应的副产物。由于选择的样品具有较强的代表性,由此推测格氏试剂与邻氯苯甲腈反应为国内非法工厂生产邻氯苯基环戊酮普遍采用的工艺,邻氯苯甲酰氯与环戊烯的傅克酰基化反应的合成方法由于需低温反应设备条件,极少被非法加工厂采用。
