非衍生化高效液相色谱-串联质谱法快速检测生物体液中草甘膦、草铵膦及代谢物
2021-05-17张云峰任昕昕王爱华董林沛吴小军张景然刘冰洁
张云峰,赵 森,常 靖,任昕昕,王爱华,赵 鹏,董林沛,吴小军,张景然,刘冰洁
(1.公安部物证鉴定中心,北京 100038;2.浙江警察学院 浙江省毒品防控技术研究重点实验室,浙江 杭州 310053;3.爱博才思亚太应用支持中心,北京 100015)
草甘膦和草铵膦是一类非选择性、内吸型除草剂,在我国农业、林业中应用广泛,其生产量和使用量均位于世界前列[1]。草甘膦和草铵膦的毒性不大,但具有致癌风险,因此受到社会越来越多的关注。此外,投毒、误服、自杀等原因的中毒(案)事件也时有发生[2-3],因此草甘膦和草铵膦在涉毒案件的检验鉴定工作中需求较大。建立生物体液(血液、尿液)中此类除草剂的检测方法可为案件的定性和侦破提供技术支持,为临床毒物分析及救治提供参考依据[4]。
草甘膦和草铵膦进入人体后会发生代谢,草甘膦的代谢产物为氨甲基膦酸、N-乙酰草甘膦、N-甲基草甘膦;草铵膦的代谢产物为N-乙酰氨甲基磷酸、3-甲基磷酸亚基丙酸、N-乙酰草铵膦[5-6]。草甘膦、草铵膦及两者的代谢物均为强极性化合物,易溶于水。目前血液和尿液中草甘膦、草铵膦及代谢物的检测方法主要有分光光度法[7]、高效液相色谱法[8]、气相色谱法[9]、气相色谱-质谱法[10]、离子色谱法[11]、离子色谱-质谱法[12]、高效液相色谱-串联质谱法(HPLC-MS/MS)[13]等。其中,分光光度法的专属性差,难以满足毒物分析特异性强、定性准确的要求;高效液相色谱法、气相色谱法、气相色谱-质谱法需进行衍生,操作复杂、不易控制、重现性差;离子色谱法易受生物基质影响,重现性差,适用范围窄。HPLC-MS/MS法具有灵敏度高、分析范围广、分析时间短等特点,近年来广泛应用于毒物分析领域[14-15]。然而,目前已报道的血液中草甘膦的HPLC-MS/MS检测方法存在灵敏度偏低、基质干扰大、样本与标准品保留时间不一致等缺点,难以满足临床及刑侦、司法鉴定中对此类除草剂的高灵敏度、简单、快速、准确的检测需求。
本研究以血液和尿液为研究对象,选择8种极性农药及代谢物,包括草甘膦及其代谢物(氨甲基膦酸、N-乙酰草甘膦、N-甲基草甘膦)、草铵膦及其代谢物(N-乙酰氨甲基磷酸、3-甲基磷酸亚基丙酸、N-乙酰草铵膦),采用非衍生化方法,建立了生物体液中草甘膦、草铵膦及代谢物等8种极性农药的HPLC-MS/MS法,为解决草甘膦、草铵膦涉毒案(事)件提供技术支持。
1 实验部分
1.1 仪器、材料与试剂
LC-30AD 高效液相色谱(日本Shimadzu公司),配有Turbo VTM离子源的Triple Quad 5500三重四极杆质谱仪(美国Sciex公司);实验用水由Milli-Q超纯水系统(美国Millipore公司)制备。高速冷冻离心机(美国ThermoFisher Scientific公司);Vortex-Genie 2可调速涡旋混合器(美国SI公司);电子分析天平(德国Sartorius公司);移液器和2 mL离心管(德国Eppendorf公司);碳酸铵和碳酸氢铵(色谱纯,Sigma公司);Cleanert PEP Plus固相萃取柱(60 mg/3 mL,天津艾杰尔科技有限公司)。
草甘膦、草铵膦、氨甲基膦酸、N-乙酰草甘膦、N-甲基草甘膦、N-乙酰氨甲基磷酸、3-甲基磷酸亚基丙酸、N-乙酰草铵膦(纯度>98%,德国LGC公司);甲醇、乙腈、甲酸、甲酸铵(HPLC级,美国Fisher Scientific);9 cm定性滤纸(杭州新华纸业有限公司)。
1.2 标准溶液的配制
准确称取各化合物固体标准品10.0 mg于10 mL容量瓶中,用水溶解并定容,得到质量浓度为1.0 mg/mL的标准储备溶液,于4 ℃避光储存。标准工作溶液由标准储备溶液用纯水稀释得到,现用现配。
1.3 样品前处理
取0.2 mL样品,加入0.2 mL水,混匀后加入0.6 mL乙腈,振荡混匀,冷冻离心10 min,取上清液,依次过Cleanert PEP Plus固相萃取柱和0.22 μm有机微孔滤膜,收集滤液于样品瓶中,待分析。
1.4 液相色谱条件
色谱柱:Metrosep A Supp 5阴离子色谱柱(150 mm×4.0 mm,5 μm);流动相:纯水(A)和200 mmol/L碳酸氢铵溶液(含0.1%氨水)(B);流速为0.6 mL/min;柱温为40 ℃;进样量为5 μL。梯度洗脱程序:0~0.5 min,10% B;0.5~1.0 min,10%~40% B;1.0~4.0 min,40%~80% B;4.0~5.0 min,80%~82.5% B;5.0~6.0 min,82.5%~95% B;6.0~7.0 min,95% B;7.0~7.1 min,95%~10%B;7.1~10.0 min,10% B。
1.5 质谱条件
离子源为电喷雾电离负离子模式(ESI-);检测方式:多反应监测(MRM);离子源温度为600 ℃;喷雾电压为- 4 500 V;雾化气(GS1)为55 psi(379.223 kPa),气帘气(CUR)为35 psi(242.325 kPa);辅助气(GS2)为45 psi(310.275 kPa);碰撞气(CAD)为High。所有分析物的保留时间、定量与定性离子对、去簇电压(DP)及碰撞能量(CE)见表1。
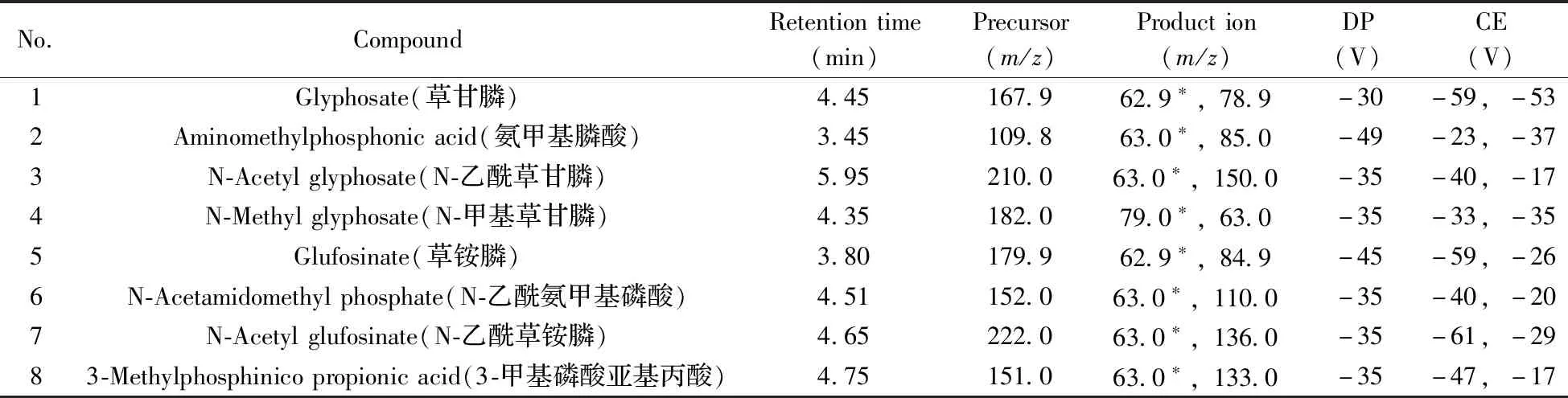
表1 8种极性农药的质谱条件Table 1 MS parameters of 8 polar pesticides
2 结果与讨论
2.1 质谱条件的优化
在电喷雾离子源正、负离子模式下,先用针泵进样分析各目标化合物的单个标准溶液,使用一级全扫描确定母离子;再采用Product Ion Scan,设定碰撞能量初始值为5 eV,以5 eV为步长自动调节碰撞能量,得到2个响应值高的特征子离子。进一步优化去簇电压和碰撞能量,最终确定8种极性农药的质谱检测条件见表1。
2.2 色谱条件的优化
草甘膦和草铵膦属于强极性农药,在常规反相色谱柱上保留较弱。实验比较了3种适用于强极性化合物的色谱柱,分别为Metrosep A Supp 5阴离子色谱柱(150 mm×4.0 mm,5 μm)、Acclaim Trinity Q1(100 mm×3.0 mm,3 μm)和Torus DEA(100 mm×2.1 mm,1.7 μm)。实验结果表明,Metrosep A Supp 5阴离子色谱柱对8种极性农药的保留时间适宜,色谱峰形良好。
在Metrosep A Supp 5阴离子色谱柱上,以纯水为流动相A,比较了不同碳酸氢铵浓度(50、100、200、250 mmol/L)和氨水含量(0.01%、0.05%、0.1%和0.2%)的流动相体系对8种极性农药色谱行为的影响。结果表明,以200 mmol/L碳酸氢铵和0.1%氨水作为流动相B时,8种极性农药的色谱峰形良好,质谱响应值高,保留时间重现性好。综上,本文选择以Metrosep A Supp 5为色谱柱,200 mmol/L碳酸氢铵溶液(含0.1%氨水)和纯水为流动相体系,得到8种极性农药标准品的总离子流图(TIC)及MRM色谱图见图1。

图1 8种极性农药标准品的总离子流图及MRM色谱图(5 ng/mL)Fig.1 Total ion and MRM chromatograms of 8 polar pesticides standard solutions(5 ng/mL)
2.3 前处理条件的优化
为保证检测结果的时效性,对血液和尿液等生物体液的前处理多采用有机溶剂蛋白沉淀法。实验比较了甲醇和乙腈的蛋白沉淀效果,结果表明,当样品直接使用有机溶剂进行蛋白沉淀时,8种极性农药的回收率均偏低。这是由于目标物的极性较强,样品粘稠,当有机溶剂比例较高时,影响了目标物的提取效果,因此在蛋白沉淀前先加入纯水对样品进行稀释。稀释后的样品,用乙腈的蛋白沉淀效果明显优于甲醇,故本文选择乙腈作为蛋白沉淀剂。
血液样品经乙腈沉淀蛋白,冷冻高速离心后,上清液会变浑浊,可能是低温下脂类杂质在溶剂中析出所致。PEP Plus固相萃取柱对脂类物质有很好的吸附效果,且无需活化,操作简单。实验结果表明,上清液通过该柱后变为澄清透明的状态。优化的前处理条件如“1.3”所示。
2.4 基质效应
取0.2 mL血液或尿液样品,按照“1.3”步骤进行提取,取上清液作为标准溶液稀释液,选择10、100 ng/mL 2个浓度水平采用本方法进行测定,并与“1.2”相应质量浓度标准溶液的峰面积作比[16]。得出所有目标农药的基质效应为86.5%~106%(见表2),表明空白血液和尿液基质对8种农药的检测影响较小,本方法可用于血液、尿液中8种农药的定量检测。
2.5 线性关系、检出限与定量下限
使用空白血液样品的净化液,分别配制质量浓度为0.5、1、2、5、10、20、50 ng/mL的基质标准溶液,进样检测。以8种目标物的质量浓度为横坐标(x,ng/mL),所得峰面积为纵坐标(y)进行线性回归。并以信噪比S/N≥3时的质量浓度为检出限(LOD),S/N≥10时的质量浓度为定量下限(LOQ)。结果显示,8种目标物在0.5~50 ng/mL范围内线性关系良好(r2>0.99),LOD为0.08~0.3 ng/mL,LOQ为0.3~1 ng/mL(见表2)。

表2 8种极性农药的线性回归方程、相关系数、基质效应、检出限和定量下限Table 2 Regression equations,correlation coefficients(r2),matrix effects,limits of detection(LOD) and limits of quantitation(LOQ) for 8 polar pesticides
2.6 回收率及相对标准偏差
取0.2 mL样品,加入适量8种农药混合标准溶液,使其样品中质量浓度为100 ng/mL。按照“1.3”步骤进行提取,每个浓度平行6份,每日早、中、晚分别测定6次,连续测定6 d。分别求得血液、尿液样品的平均回收率以及日内、日间相对标准偏差(RSD),结果见表3。由表3可知,血液和尿液中8种极性农药的加标回收率为81.5%~114%,RSD为1.1%~5.0%;日内及日间RSD分别为0.30%~2.8%和0.50%~5.3%。实验结果良好,满足法庭科学检验标准。

表3 8种极性农药的相对标准偏差及回收率(n=6)Table 3 Relative standard deviations(RSD) and recoveries of 8 polar pesticides(n=6)
2.7 实际案例应用
2020年8月,吉林省某女子因家庭矛盾将其丈夫杀害,归案后供述曾购买除草剂服用,企图自杀。除草剂名称为“草*膦”,办案单位提取了该女子的血液送检。取0.2 mL血液,按照本方法进行分析,结果检出了草甘膦及其代谢物氨甲基膦酸、N-乙酰草甘膦和N-甲基草甘膦,其色谱图见图2。结果表明,本研究建立的方法可用于实际案例的检验。
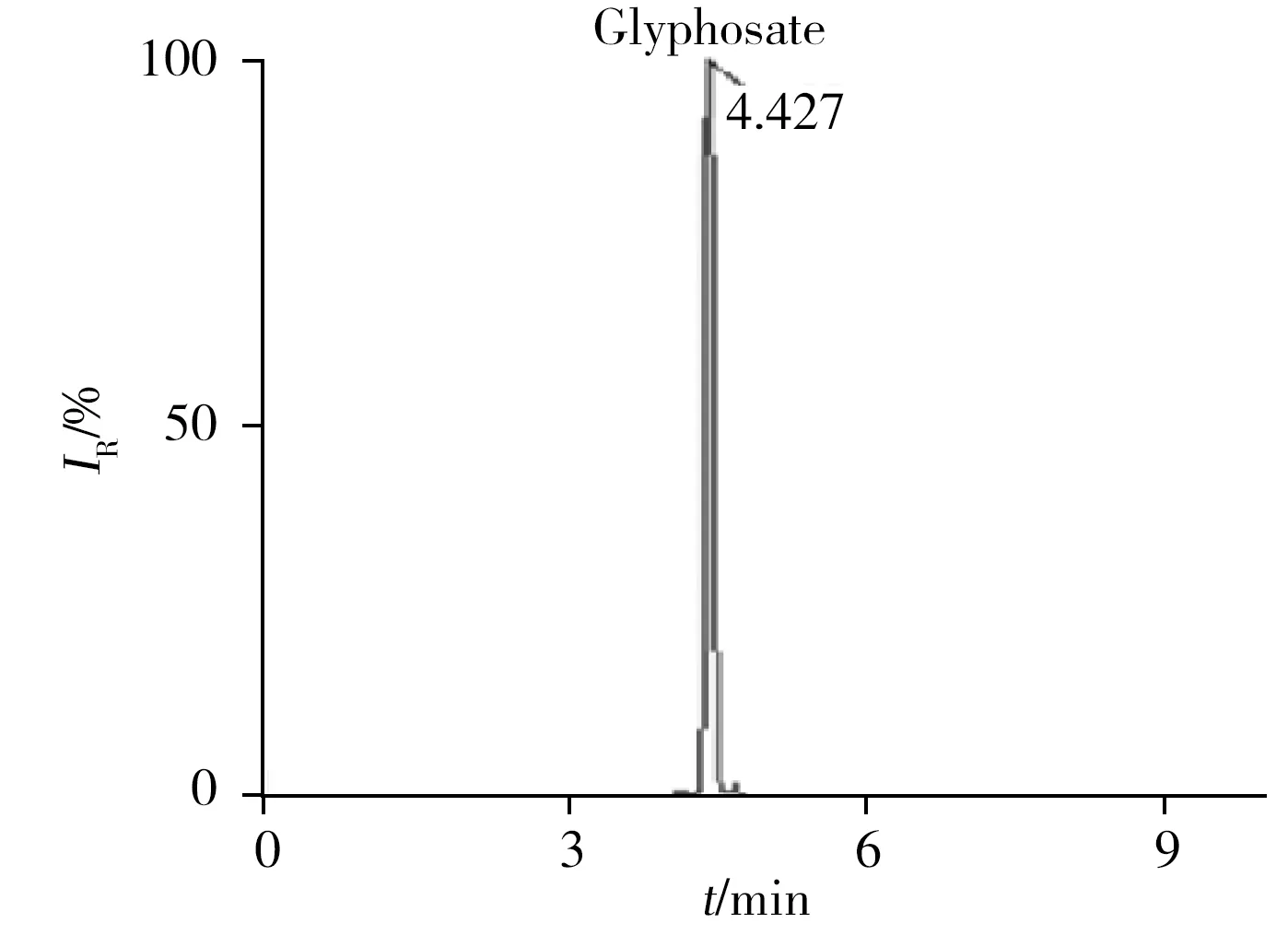
图2 血液中检出的草甘膦及其代谢物色谱图Fig.2 Chromatograms of glyphosate and its metabolites in blood sample
3 结 论
本研究建立了血液和尿液中草甘膦、草铵膦等8种极性农药的非衍生化HPLC-MS/MS检测方法。与衍生化方法相比,该方法具有操作简便、省时省力、灵敏度高、准确性好、重现性好等优点,克服了以往LC-MS/MS检测草甘膦、草铵膦稳定性和重现性差的缺点,能够快速、准确地实现对生物体液中草甘膦、草铵膦等8种极性农药的定性定量分析,满足草甘膦、草铵膦涉毒案(事)件的检验鉴定要求。
