CoMoSx/CeO2-γ-Al2O3催化剂的制备及其催化还原SO2的性能
2021-05-16矫庆泽耿云峰黎汉生史大昕冯彩虹
任 健 矫庆泽,2 王 彤 耿云峰 黎汉生*,吴 芹*, 史大昕 赵 芸 冯彩虹
(1北京理工大学化学与化工学院,化学电源与绿色催化北京市重点实验室,北京 100081)
(2北京理工大学珠海学院化工与材料学院,珠海 519085)
(3北京北大先锋科技有限公司,北京 100080)
0 引 言
燃煤烟气中的SO2是大气污染物的主要来源之一,煤烟气脱硫技术研究已引起世界各国的广泛关注。利用还原剂将烟气中分离出来的SO2还原为单质硫的技术不仅可以降低SO2对环境的污染,产生的硫磺还可以缓解我国硫资源匮乏的现状[1],且过程中无需消耗水,也不会产生固体废物,具有较大的应用前景[2]。CO具有来源广泛、副反应少、产物纯度高等优点,被认为是具有工业前景的还原剂[3]。因此,利用催化剂催化CO还原SO2生产硫单质技术具有较好的应用前景。
目前CO还原SO2制硫单质的催化剂按照催化反应机理分为3大类:COS中间产物机理、氧化还原机理、协同作用机理。遵循COS中间产物机理的催化剂以过渡金属硫化物为活性组分,CO与硫化物反应产生的COS具有较强的还原性,更容易促进SO2的还原,因而具有较高的催化活性和选择性[4-6]。但在反应温度较低时COS与SO2的反应速率较慢,金属硫化物表面的S以COS的形式流失,既影响催化剂的使用寿命,又对大气环境构成威胁。遵循氧化还原机理的催化剂以CeO2、TiO2及其复合金属氧化物为主,氧化物晶格氧的迁移使催化剂具有良好的氧化还原性能[7-9]。催化剂在反应过程中不会产生COS,但是氧空位的形成及移动需要较高的温度,因此金属氧化物催化SO2还原制单质硫的反应温度高。遵循协同作用机理的催化剂以金属硫化物和氧空位为活性中心,CO与金属硫化物反应产生的COS可在较低温度下促进氧化物表面氧空位的产生,因而提高了低温下的反应活性并促进了COS的转化[10-11]。协同作用机理的催化剂具有反应温度较低、产物选择性高等优势,具有广阔的工业应用前景。
本课题组前期研究表明[12],CoMoSx/γ-Al2O3催化剂遵循COS中间产物机理,具有优异的催化性能,反应温度400℃时,SO2的转化率、硫单质的选择性和产率都接近100%,但是反应温度较高、能耗高。如何进一步改进CoMoSx/γ-Al2O3催化剂,对于CO还原SO2制硫单质体系具有重要意义。协同作用机理的优势为催化剂的改进提供了思路:CeO2具有较高的储放氧的能力、独特的氧化还原特性、优异的抗硫性能,其氧空位具有较高的迁移性[13],利用CeO2助剂对CoMoSx/γ-Al2O3催化剂改性,使催化剂具有金属硫化物和氧空位的双活性中心,将有助于提高催化剂的催化性能。
我们以少量的CeO2为助剂对γ-Al2O3载体进行改性,采用等容浸渍法制备了CoMoSx/CeO2-γ-Al2O3催化剂。采用X射线衍射(XRD)、高角环形暗场像-扫描透射电子显微镜(HAADF-STEM)、X射线光电子能谱(XPS)、程序升温脱附/程序升温还原/程序升温表面反应(TPD/TPR/TPSR)等方法对其进行表征,探究CeO2助剂及助剂用量对反应温度及催化性能的影响;研究了CoMoSx/CeO2-γ-Al2O3催化剂在CO还原SO2的反应中的催化活性及选择性;分析催化剂的还原性及其对SO2和中间产物COS的吸附能力,并对其催化反应机理进行探讨,考察了催化剂的稳定性。
1 实验部分
1.1 催化剂的制备
称取 0.851 4 g Ce(NO3)3·6H2O(分析纯,国药集团化学试剂有限公司),溶解于5.6 m L去离子水中形成均质溶液。将10 gγ-Al2O3载体(山东瑞鼎科技有限公司)等体积浸渍于该溶液中,充分浸渍12 h后在110℃下干燥12 h除去水分,在520℃下煅烧12 h,得到CeO2物质的量分数为1%的CeO2-γ-Al2O3改性载体。然后依次称取一定量的(NH4)6Mo7O24·4H2O(分析纯,国药集团化学试剂有限公司)和Co(NO3)2·6H2O(分析纯,国药集团化学试剂有限公司),溶解于去离子水中形成均质溶液后对载体进行等体积浸渍,经相同的干燥、煅烧程序后得到氧化物催化剂4.4% COO-16% MoO3/CeO2-γ-Al2O3(CoO和MoO3的含量均为质量分数),即 CoMoO4/CeO2-γ-Al2O3。最后对氧化物催化剂进行预硫化:以氩气为保护气,将催化剂在H2S体积分数10%的H2S/H2的混合气中于500℃预硫化1 h,得到预硫化后的催化剂CoMoSx/CeO2-γ-Al2O3。制备了不同 CeO2添加量(物质的量分数)的催化剂,分别标记为CoMoSx/1.0% CeO2-γ-Al2O3、CoMoSx/1.5% CeO2-γ-Al2O3和CoMoSx/2.0% CeO2-γ-Al2O3。
1.2 催化剂的表征
对载体和CoMoSx/1.5% CeO2-γ-Al2O3催化剂进行了各项表征。XRD测试在Rigaku公司生产的Ultima Ⅳ型X射线衍射仪上进行,辐射源为CuKα(λ=0.154 nm),工作电压40 kV,工作电流40 mA,扫描范围 2θ=10°~80°,扫描速度 10(°)·min-1。采用FEI公司生产的TF20型透射电子显微镜,选取200 kV的加速电压,对催化剂的形貌及表面元素的分布情况进行了HAADF-STEM表征。采用ULVAC-PHI公司生产的PHI QUANTERA-Ⅱ SXM型X射线光电子能谱仪分析硫化后的催化剂中Mo和Co的结合能,选取AlKα靶(hν=1 486.6 eV),功率300 W,以无定形碳(C1s为284.6 eV)的电子结合能作为校正标准,对Co、Mo元素进行测试。
TPR、TPD和TPSR在天津先权公司生产的化学动态吸附仪(TP-5076)上进行测试,用Hiden公司的HPR-20型四级杆质谱对出口气体进行定量分析。TPR:样品装填量为0.1 g,以CO体积分数5%的CO/Ar为还原气,总流量为50 mL·min-1,以10 ℃·min-1的速率将样品加热至700℃,记录CO2和COS的信号曲线变化情况。TPD:样品装填量为0.1 g,在室温下分别通入流量为50 m L·min-1的SO2体积分数5%的SO2/Ar和COS体积分数5%的COS/Ar,待催化剂吸附饱和后用氩气吹扫反应器,然后在氩气气氛下以10℃·min-1的速率将样品加热至700℃,记录SO2和COS的脱附情况。TPSR:样品装填量为0.1 g,在室温下通入流量为50 mL·min-1的SO2体积分数5%的SO2/Ar,样品吸附饱和后用氩气吹扫反应器,然后通入50 mL·min-1的CO体积分数5%的CO/Ar,在该气氛下以10℃·min-1的速率将样品加热至700℃,记录不同温度下尾气中各组分的含量变化。
1.3 催化剂的性能评价


2 结果与讨论
2.1 CoMoS x/CeO2-γ-Al2O3催化剂的表征
图1 为γ-Al2O3、CeO2-γ-Al2O3、CoMoO4/CeO2-γ-Al2O3及CoMoSx/CeO2-γ-Al2O3催化剂的XRD 图。图中2θ=37.60°、45.79°和66.76°处的峰对应γ-Al2O3的衍射峰(PDF No.29-0063)[15]。CeO2-γ-Al2O3、CoMoO4/CeO2-γ-Al2O3与γ-Al2O3载体的衍射峰基本相同,没有出现CeO2、CoO和MoO3的衍射峰,这是因为γ-Al2O3会与多种氧化物物种发生强相互作用,导致负载的氧化物物种高度分散,进而观察不到其特征衍射峰。预硫化后的CoMoSx/CeO2-γ-Al2O3催化剂仍然保持γ-Al2O3的主体结构,在 2θ=33.3°、58.3°和60.4°出现了MoS2的衍射峰,没有出现其他的衍射峰,催化剂经预硫化后没有发生团聚。

图1 载体和催化剂的XRD图Fig.1 XRD patterns of the supports and catalysts
图2 是 CoMoSx/CeO2-γ-Al2O3催化剂的HAADFSTEM图。由图2a可知催化剂为无定形状态,由图2b可知Co、Mo、Ce元素均匀分散在催化剂表面,没有发生团聚,说明采用等体积浸渍法制备的催化剂具有较好的分散性。

图2 CoMoS x/CeO2-γ-Al2O3催化剂的HAADF-STEM图Fig.2 HAADF-STEM images of CoMoS x/CeO2-γ-Al2O3
为了更好地了解催化剂表面金属元素的化学状态,对催化剂进行了XPS分析。图3a为预硫化的CoMoSx/CeO2-γ-Al2O3催化剂上Mo3d的XPS谱图,其中电子结合能为228.6和231.7 eV处的谱峰分别归属于 Mo4+3d5/2和 Mo4+3d3/2,对应 MoS2和 CoMoS;电子结合能为233.0和235.4 eV处的谱峰分别归属于Mo6+3d5/2和 Mo6+3d3/2,对应未被硫化的 MoO3;电子结合能为232.1和233.1 eV处的谱峰归属于Mo5+3d5/2和 Mo5+3d3/2,对 应 Mo2O5和 MoOxSy[16]。 由 表 1 可 知CoMoSx/CeO2-γ-Al2O3催化剂中Mo的硫化物所占比例约为84.4%,而CoMoSx/γ-Al2O3催化剂中Mo的硫化物约为57.9%,硫化程度提高了约1.5倍,CeO2的改性削弱了活性组分与载体之间的相互作用,使催化剂上的MoO3更容易被硫化[17-18]。图3b为预硫化的CoMoSx/CeO2-γ-Al2O3催化剂上 Co2p3/2的 XPS谱图。电子结合能为778.5 eV处的谱峰归属于Co—Mo—S活性相;电子结合能为781.2 eV处的谱峰对应于氧化态的Co,可能是样品转移过程中Co被空气中的氧气氧化导致。

图3 CoMoS x/CeO2-γ-Al2O3的 Mo3d(a)和Co2p(b)的XPS谱图Fig.3 XPS spectra of Mo3d(a)and Co2p(b)of CoMoSx/CeO2-γ-Al2O3

表1 CoMoS x/CeO2-γ-A l2O3和CoMoS x/γ-A l2O3[12]的M o3d能谱分析结果对比Table 1 Comparison of M o3d energy spectrum analysis results of CoMoS x/CeO2-γ-A l2O3 and CoMoS x/γ-A l2O3[12]
2.2 CoMoS x/CeO2-γ-Al2O3催化剂的 TPD/TPR/TPSR分析
图4是γ-Al2O3载体、CeO2-γ-Al2O3载体、CoMoSx/γ-Al2O3催化剂、CoMoSx/CeO2-γ-Al2O3催化剂在程序升温条件下的SO2脱附图及CoMoSx/CeO2-γ-Al2O3催化剂在氩气气氛下加热分解产生的SO2信号图。从图中可以看出,与γ-Al2O3载体相比,CeO2-γ-Al2O3改性载体的SO2脱附峰面积减小,说明CeO2的改性覆盖了γ-Al2O3载体表面部分SO2吸附位,使SO2的吸附量减少。升温过程中CoMoSx/CeO2-γ-Al2O3催化剂在70和520℃左右分别出现SO2的信号峰,第一个峰对应SO2在载体上的吸附,由峰强度可知SO2的吸附量大大减少,这是由于CoMoSx的负载覆盖了载体上大部分的SO2吸附位点,导致SO2的吸附量减少;SO2的第二个峰与催化剂在氩气气氛下分解产生的SO2信号相似,但峰面积大于催化剂分解产生的SO2信号峰面积,说明SO2的第二个峰大部分由催化剂的分解产生,一小部分由吸附在CoMoSx上的SO2脱附产生。CoMoSx/γ-Al2O3催化剂的第一个脱附峰的面积小于 CoMoSx/CeO2-γ-Al2O3催化剂,说明 CeO2对活性组分与载体的削弱作用可以使催化剂吸附更多的 SO2;CoMoSx/γ-Al2O3催化剂的第二个脱附峰在420℃左右,即催化剂的分解温度约为420℃[12],而经CeO2改性后的CoMoSx/CeO2-γ-Al2O3催化剂的分解温度约为520℃,说明CeO2的引入提高了催化剂的分解温度。

图4 载体和催化剂的SO2-TPD及CoMoS x/CeO2-γ-Al2O3催化剂的热分解图Fig.4 SO2-TPD of the supports and catalysts and thermal decomposition of CoMoS x/CeO2-γ-Al2O3
图5 为γ-Al2O3载体、CeO2-γ-Al2O3、CoMoSx/γ-Al2O3催化剂、CoMoSx/CeO2-γ-Al2O3催化剂在程序升温条件下的COS脱附图。与γ-Al2O3载体相比,CeO2的改性对COS在载体上的吸附几乎没有影响,根据COS脱附峰的位置和信号强度可知COS的吸附属于弱吸附作用且吸附量很少。与CoMoSx/γ-Al2O3催化剂相比[12],CoMoSx/CeO2-γ-Al2O3催化剂出现了一个与载体对应的COS脱附峰,说明催化剂对COS的吸附归因于载体对COS的吸附,CoMoSx不会吸附COS。由于CoMoSx覆盖了载体上的COS吸附位点,CoMoSx/CeO2-γ-Al2O3催化剂只吸附微量的COS气体。
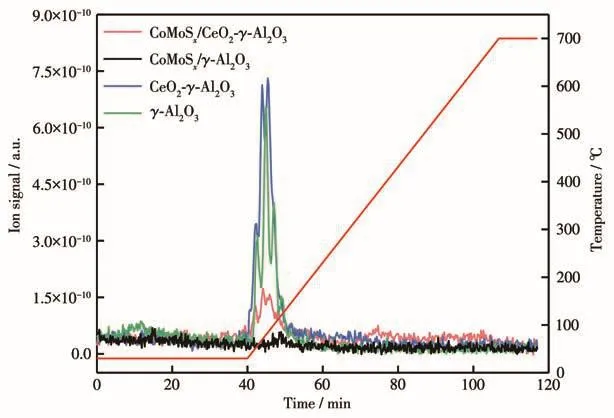
图5 载体和催化剂的COS-TPD图Fig.5 COS-TPD of the supports and catalysts
图6 为 CoMoSx/CeO2-γ-Al2O3和 CoMoSx/γ-Al2O3催化剂的CO-TPR图。图中可看出CO还原催化剂的过程中出现了COS和CO2的特征峰,说明CO经过程序升温还原生成了COS和CO2,分析认为COS是由CO与催化剂表面的金属硫化物反应产生的[19],CO2信号峰的出现一方面是由于催化剂的不完全硫化,另一方面可能是由CeO2还原产生。与CoMoSx/γ-Al2O3催化剂相比,CoMoSx/CeO2-γ-Al2O3催化剂的COS和CO2的出峰温度较低,说明CO可在更低温度下还原 CoMoSx/CeO2-γ-Al2O3,即 CeO2改性提高了催化剂的还原性。CO2信号在170~250℃之间出现了一个较高的尖峰,这是由于COS与CeO2中高迁移性的晶格氧发生反应,从而在较低温度下生成了CO2[20]。CoMoSx/CeO2-γ-Al2O3催化剂的 COS 信号值远高于CoMoSx/γ-Al2O3催化剂,而CO2的信号值较低,由XPS分析结果可知,CoMoSx/CeO2-γ-Al2O3催化剂的硫化程度更高,催化剂表面金属硫化物的含量更高,因而CO还原催化剂产生的COS量增多,CO2的量减少。

图6 CoMoS x/CeO2-γ-Al2O3和CoMoS x/γ-Al2O3催化剂的CO-TPR图Fig.6 CO-TPR of CoMoS x/CeO2-γ-Al2O3 and CoMoS x/γ-Al2O3
图7 是 CoMoSx/CeO2-γ-Al2O3和 CoMoSx/γ-Al2O3催化剂的(SO2+CO)-TPSR图。与催化剂的SO2-TPD、CO-TPR 相比,CoMoSx/γ-Al2O3催化剂的 SO2的第一个脱附峰的强度变化很小,COS的信号峰强度几乎不变,说明反应温度低于250℃时SO2与COS的反应速率较慢;温度高于300℃时,SO2的第二个峰几乎消失,CO2的信号值迅速增大,说明SO2的转化率升高。而CoMoSx/CeO2-γ-Al2O3催化剂的SO2的第一个脱附峰强度略有降低,COS的信号峰强度减少,说明在低温下SO2和COS发生反应,由于温度较低时COS与SO2反应的速率较慢,分析认为COS和SO2的消耗是由于强还原性的COS在较低温度下还原催化剂表面的CeO2,产生了氧空位和CO2,在氧空位的迁移下部分SO2被还原,导致SO2和COS的信号强度均降低,验证了CO-TPR中对CO2尖峰的分析。SO2的第二个峰几乎完全消失,说明这部分的SO2被完全消耗,由此推断催化剂的最佳反应温度在250℃以上。与CoMoSx/γ-Al2O3催化剂相比,CoMoSx/CeO2-γ-Al2O3催化剂CO2信号的出峰温度较低,说明CeO2的引入有助于促进COS和SO2在较低温度下反应产生CO2。

图7 CoMoS x/CeO2-γ-Al2O3和CoMoS x/γ-Al2O3的(SO2+CO)-TPSR图Fig.7 (SO2+CO)-TPSR of CoMoS x/CeO2-γ-Al2O3 and CoMoS x/γ-Al2O3
由以上分析可知,CeO2削弱了载体与金属氧化物之间的相互作用,提高了CoMoSx/CeO2-γ-Al2O3催化剂的硫化程度,使COS的生成量增多;COS在较低温度下还原CeO2,可促进COS的转化,提高S单质的选择性,且CeO2还原产生的氧空位有助于提高SO2的转化率,即CoMoSx/CeO2-γ-Al2O3催化剂遵循COS与氧空位协同作用的机理,推测其机理为:

其中,□表示硫缺位,[]表示晶格氧空位。
该机理与Kim等[20]的协同作用机理一致,说明CeO2助剂的引入使CoMoSx/CeO2-γ-Al2O3催化剂按照协同机理催化反应过程。
2.3 CoMoS x/CeO2-γ-Al2O3催化剂的催化性能
图8为不同CeO2添加量的CoMoSx/CeO2-γ-Al2O3催化剂在不同温度下催化CO还原SO2的催化活性。由图8可知,与CoMoSx/γ-Al2O3催化剂相比,CeO2的引入使催化剂的活性明显提高,这是由于Co—Mo—S活性位和CeO2的氧空位协同作用,促进了COS与SO2的反应。由图8a可知,随着CeO2含量的增加,相同温度下SO2的转化率先升高后降低,CeO2的添加量为1.5%时,SO2的转化率最高,由于CeO2含量过高不利于COS与SO2的直接反应,因此CoMoSx/2.0% CeO2-γ-Al2O3SO2催化SO2的转化率降低。随着温度的升高,SO2的转化率逐渐升高,反应温度为350℃时CoMoSx/1.5% CeO2-γ-Al2O3催化剂的SO2转化率可达99%以上。SO2的转化率与反应温度呈正相关,说明高温有利于SO2的转化。由图8b、8c可知,CoMoSx/CeO2-γ-Al2O3催化剂的COS选择性随反应温度的升高而降低,S单质的选择性随温度的升高而升高,进一步说明了高温有利于促进COS与SO2的反应。CoMoSx/1.5% CeO2-γ-Al2O3催化剂在不同温度下的S选择性均高于CoMoSx/γ-Al2O3催化剂。反应温度高于350℃时,CoMoSx/γ-Al2O3催化剂的S的选择性约为 94%[12],而 CoMoSx/CeO2-γ-Al2O3催化剂的 S的选择性均可达到99%以上。由图8d可知,S的产率随反应温度的升高而升高。当反应温度低于300℃时,由于COS与SO2的反应速率较慢,S的产率较低。反应温度为300℃时,CoMoSx/γ-Al2O3催化剂的S产率约为16%[12],而CoMoSx/CeO2-γ-Al2O3催化剂的 S 产 率远高于 CoMoSx/γ-Al2O3,其 中 CoMoSx/1.5% CeO2-γ-Al2O3催化剂的S产率约为76%,为CoMoSx/γ-Al2O3催化剂的4.8倍。反应温度升高至350 ℃时,CoMoSx/CeO2-γ-Al2O3催化剂的S产率均可达到98%以上,其中CoMoSx/1.5% CeO2-γ-Al2O3催化剂的S产率达到了99%,SO2几乎完全转化为S单质。实验结果与2.2节的协同催化机理分析结果一致,进一步说明了协同催化机理分析的可靠性。由以上分析可知,CeO2添加量为1.5%左右时的CoMoSx/CeO2-γ-Al2O3催化剂具有最佳的催化活性。

图8 不同CeO2添加量的CoMoS x/CeO2-γ-Al2O3催化剂的催化活性Fig.8 Catalytic activity of CoMoS x/CeO2-γ-Al2O3 catalysts with different CeO2 addition amounts
2.4 CoMoS x/CeO2-γ-A l2O3催化剂的稳定性分析
为了研究改性催化剂的稳定性,考察了CoMoSx/1.5% CeO2-γ-Al2O3催化剂在300℃连续反应过程中催化活性的变化情况,结果如图9所示。从图中可以看出,SO2的转化率稳定在88%左右,S的选择性约为86%,COS的选择性约为14%,S的产率稳定在75%以上,在连续反应的200 h内,催化剂的活性没有发生明显变化,说明CoMoSx/CeO2-γ-Al2O3催化剂具有较好的稳定性,具有较好的工业化应用前景。
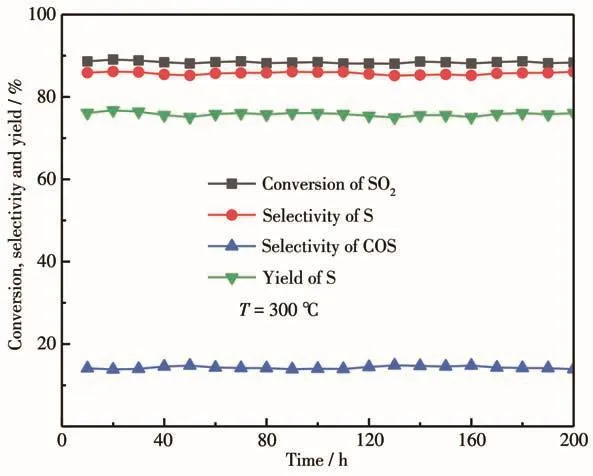
图9 CoMoS x/CeO2-γ-Al2O3催化剂的性能随反应时间变化图Fig.9 Catalytic performance of CoMoS x/CeO2-γ-Al2O3 varying with reaction time
3 结 论
以少量CeO2为助剂对γ-Al2O3载体进行改性,通过等容浸渍法制备了不同CeO2助剂负载量的CoMoSx/CeO2-γ-Al2O3催化剂。采用 XRD、HAADFSTEM、XPS、TPR、TPD、TPSR对其进行表征,研究了其催化CO还原SO2制单质硫的性能。结果表明:(1)等容浸渍法制备的催化剂具有良好的分散性;(2)CeO2改性削弱了载体与活性组分之间的相互作用,提高了催化剂的硫化程度;(3)CoMoSx/CeO2-γ-Al2O3催化剂可以吸附更多的SO2,几乎不吸附COS;(4)CeO2改性提高了催化剂的低温还原性;(5)COS可以在较低温度下还原CeO2,提高了低温下SO2的转化率和S单质的选择性,使催化剂按照协同作用机理进行;(6)CeO2助剂的改性降低了反应温度,使CoMoSx/CeO2-γ-Al2O3具有优异的催化活性,CeO2物质的量分数为1.5%的CoMoSx/CeO2-γ-Al2O3催化剂具有最佳的催化活性,350℃时SO2的转化率和S的产率可达99%以上,催化剂在300℃连续运行200 h仍保持稳定的催化性能。
