头皮上皮样血管瘤一例
2021-05-11陈佳玲陈嵘祎谢嘉豪史建强
陈佳玲 陈嵘祎 谢嘉豪 史建强
1广东医科大学附属医院皮肤科,广东湛江,524001;2南方医科大学皮肤病医院(广东省皮肤病医院),广东广州,510091
临床资料患者,女,36岁。头皮丘疹、结节3年余。3年前无名显诱因头皮出现绿豆至黄豆大红色丘疹、结节,无伴瘙痒、疼痛,未予诊治。皮疹增多并缓慢增大呈半球形,未见明显融合,无破溃及出血。患者既往体健,无外伤史,无不良嗜好,家族中无类似疾病史。体格检查:生命体征平稳,全身浅表淋巴结未触及肿大,各系统检查未见明显异常。皮肤科情况:头皮可见数个黄豆大半球形红褐色结节,质韧,边界清楚,互不融合,表明光滑,偶见搔抓后破溃,无糜烂渗出,无明显压痛(图1a)。头皮组织病理示:真皮内和皮下组织见大量血管腔隙组成的小叶状团块,境界不清,增生的血管内皮细胞肥大而圆,突向管腔,间质可见淋巴组织和大量嗜酸粒细胞浸润(图 2a~2c)。诊断:上皮样血管瘤。治疗:患者拒绝手术,给予得宝松(复方倍他米松)皮损内注射联合复方丙酸氯倍他索软膏外用,治疗一周后皮疹稍消退,趋于扁平(图1b),现已失访。
讨论上皮样血管瘤(epithelioid hemangioma, EH)又称血管淋巴增生伴嗜酸性粒细胞浸润(angiolymphoid hyperplasia with eosinophilia, ALHE),是一种不常见的良性皮肤或皮下肿瘤,为各种刺激下不成熟血管和慢性炎症细胞的局限性混合增生,炎症细胞中常有嗜酸粒细胞[1]。表现为真皮和皮下组织的红棕色或粉色无痛性血管结节,组织细胞样内皮细胞增生、明显的淋巴和嗜酸粒细胞浸润,偶见外周血嗜酸粒细胞增多[2]。病因尚不明确,可能与皮下动静脉分流继发的血管畸形、创伤、感染(HTLV或疱疹病毒8)、激素分泌失调、放疗及接触砷等有关[1]。
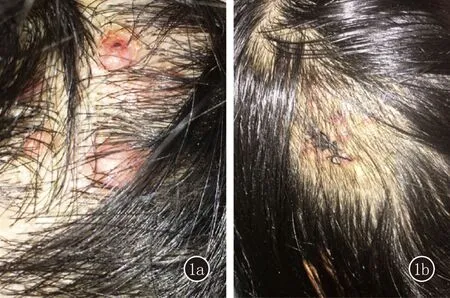
图1 1a:治疗前;1b:治疗后

图2 2a:真皮内和皮下组织见大量血管腔隙组成的小叶状团块,境界不清(HE,×40);2b:血管内皮细胞增生,间质见淋巴组织和嗜酸粒细胞浸润(HE,×100);2c:增生的血管内皮细胞肥大而圆,突向管腔(HE,×400)
本病好发于 21~50岁,女性稍多见,皮损最常发生在头颈部,其他部位例如躯干,下肢、手、阴茎、口腔黏膜和结肠很少见,常与头相关[3]。临床常表现为单发或多发、无痛的红色小丘疹或结节,或斑片[4],直径1~10 cm(0.5~3 cm常见),质韧/硬,表面光滑,可逐渐发展成红色质硬肿物,并破溃出血[5]。病变多发时,通常成团排列或呈带状分布,可以融合。一般无自觉症状,可有轻微瘙痒或触痛、搏动感。
在组织病理学上,上皮样血管瘤病变表现为真皮或皮下不同大小的血管增生团块,境界不清,血管具有肥大而圆的内皮细胞,胞浆丰富呈强嗜酸性,常显示出空泡化和细胞质进入管腔,但无多形性及有丝分裂象,部分可见细胞实性条索。血管增生与由淋巴细胞,嗜酸粒细胞和肥大细胞组成的弥漫性炎症浸润混合。有时可见有生发中心的淋巴滤泡。另外,纤维化是常见的伴随特征[6]。
结合本患者的临床表现和病理学检查诊断明确。
本病需与木村病(Kimura病)、上皮样血管瘤性结节、化脓性肉芽肿、上皮样血管内皮瘤、皮肤淋巴瘤等疾病鉴别。ALHE是血管起源的病变,木村病却是一种慢性炎症过程[3]。木村病好发于颌下腺、腮腺、耳前部位,常形成较大肿块,伴淋巴结肿大,外周血嗜酸粒细胞及IgE升高,可累及肾脏等多系统自身免疫性损害,组织病理表现血管增生不明显,无突出的管腔内皮细胞[3],可见广泛嗜酸性微脓肿、淋巴滤泡及多核细胞。皮肤上皮样血管瘤性结节常单发,多见于躯干及四肢,组织病理显示肿瘤细胞及核均较大,可见含铁血黄素沉积可沉积于瘤结节内管腔,免疫组化提示CD31、CD34 和Ⅷ因子阳性[7]。化脓性肉芽肿又称分叶状毛细血管瘤,常继发于外伤后,增长迅速,不慎触碰易出血,组织病理无明显嗜酸粒细胞浸润。
上皮样血管瘤少数情况下皮损可自然消退,最有效治疗为手术切除。切除可以局部复发(30%~40%),与病变下方没有完全切除的动静脉瘘有关。另有这些非手术常规疗法,例如糖皮质激素(局部和全身应用)、甲氨蝶呤、普萘洛尔、异维A酸,冷冻疗法,沙利度胺光疗以及针对细胞因子的新疗法,包括局部应用咪喹莫特,白介素5(mepolizumab),血管内皮生长因子A(贝伐单抗)和干扰素,激光疗法也已用于治疗ALHE病[8],但非手术疗法复发率普遍较高。
