基于气相色谱-串联质谱技术测定植物组织中糖与糖醇
2021-05-10坚乃丹李文丽张祝莉朱霞鲜学海牛伊宁
坚乃丹,李文丽,张祝莉,朱霞,鲜学海,牛伊宁*
1(甘肃省干旱生境作物学重点实验室,甘肃 兰州,730070)2(临夏市农产品质量安全中心,甘肃 临夏,731100)
糖类化合物主要是多羟基的醛或酮及其缩聚物和某些衍生物,源自植物光合作用的初级产物对生物组织结构及生命活动起着重要作用,广泛存在于自然界[1]。植物细胞中糖和糖醇作为供能代谢基质,具有调控生长、发育、繁殖和免疫等生理功能,参与植物体内三羧酸循环、糖酵解等基础代谢过程;同时作为信号分子调节相关基因的表达和酶的活性、清除多余自由基和调节植物细胞渗透压从而提高植物抗逆性[2]。因此植物与糖类互作机制及植物体内糖结构与生物学功能的相关性及其作用机理已成为新研究热点之一[3],但是植物体中低分子质量的糖与糖醇的浓度较低,所以亟需建立一种准确高效的糖与糖醇定量分析方法。
目前糖类化合物分析方法有高效毛细管电泳法、阴离子交换色谱法、高效液相色谱法、凝胶渗透色谱法、气相色谱法、质谱法及核磁共振法等[4-9]。其中气相色谱法样品用量少,分离能力强,适用于检测没有使用限量规定的糖化合物,但其只能通过保留时间进行定性和定量分析,检测结果的准确性易受复杂基质干扰[10]。质谱法具备高分辨率和高灵敏度等特点,可获得化合物的结构信息,并且推算化合物的分子质量,对于鉴别有机物分子具有重要作用[11]。气相色谱-串联质谱法(gas chromatography-tandem mass spectrometry,GC-MS/MS)兼具色谱的高分离效能与质谱高准确性的特点,特别多级质谱串联(MS/MS)技术是通过对分子离子和其被碎裂后获得的碎片离子2次扫描监测以进行数据分析,可提供清晰的分子离子与碎片离子之间的关系,增加定量准确性,广泛应用于复杂基质中痕量物质的检测[12]。丁洁等[13]利用气相色谱法测定金银花中含有的12种糖和糖醇并绘制了其指纹图谱。BOLDIZAR等[14]采用气相色谱-质谱联用法对香芹果实及叶片组织中的糖和糖醇进行定量分析。但是还尚未见文献报道使用气相色谱-串联质谱法同时测定多种糖与糖醇的方法。
1 材料与方法
1.1 材料与试剂
原料:黑果枸杞购自青海格尔木市大格勒乡;芦荟采自室内栽培植株。
试剂:吡啶、乙酸酐、盐酸羟胺均为分析纯,成都科隆化学品有限公司;色谱级氯仿,青岛天泽生物技术有限公司;侧金盏花醇(Ado)、葡萄糖(Glu)、半乳糖(Gal)、肌醇(Ino)、甘露醇(Man)、山梨醇(Sor)、蔗糖(Suc)标准品(纯度均大于98%),成都乐美天医药科技有限公司。
1.2 仪器与设备
GC-MS/MS三重四极杆气相色谱质谱联用仪7890B-7000D(配有EI离子源和NIST MS数据处理系统),美国Agilent公司;DF-110S集热式磁力搅拌器,常州瑞华制造有限公司;毛细管柱DB-1701、DB-23、HP-5MS(均为30 m × 0.25 mm,0.25 μm),美国Agilent公司;冷冻台式真空离心浓缩仪,日本京东理化公司。
1.3 标准溶液配制
精确称取侧金盏花醇2 mg,用无水吡啶溶解并定容至10 mL作为内标。另精确称取其他糖和糖醇标准品(葡萄糖、半乳糖、肌醇、甘露醇、山梨醇、蔗糖)2 mg,用无水吡啶溶解并定容至10 mL。分别吸取各标准品1.4、1.2、1.0、0.8、0.6、0.4、0.2、0.1、0.05、0.025 mL于离心管中,分别加入内标溶液0.5 mL,再以无水吡啶补充至2 mL。于4 ℃冰箱避光保存。
1.4 样品提取
1.4.1 氯仿-甲醇水溶剂提取法[15]
称取0.2 g植物样品进行液氮研磨,加入4 mL提取液(甲醇-氯仿-水体积比12∶5∶3),充分摇匀,再加入4 mL去离子水,混和均匀,离心取上清液至试管,旋转蒸干,补充2 mL吡啶,于4 ℃冰箱保存。
1.4.2 超声辅助提取法
称取0.2 g植物样品进行液氮研磨,置于20 mL的离心管,加入5 mL去离子水,10 mL的无水乙醇,置超声仪超声30 min,离心,4 000 r/min,去上清。残留不溶物用10 mL 80%的乙醇水溶液洗涤、离心,去上清,加入10 mL水,超声提取30 min,重复2次。冷却至室温,离心取上清液,旋转蒸干,补充2 mL吡啶,于4 ℃冰箱保存。
1.5 标准品及样品乙酰化处理[16]
向待测样品及标准品溶液加入20 mg盐酸羟胺充分溶解,置于90 ℃水浴锅反应30 min,冷却至室温,加入1 mL无水乙酸酐,再次于90 ℃下反应30 min,冷却后加入1 mL去离子水搅拌,然后用氯仿溶液萃取3次,合并氯仿层,旋转蒸干,加入1 mL氯仿溶解,进行分析检测。
1.6 分析条件
1.6.1 色谱条件
色谱柱:HP-5MS色谱柱(30 m×0.25 mm,0.25 μm);进样口温度:300 ℃,载气:高纯氦气;碰撞气:高纯氮气,纯度≥99.999%;柱流量:1 mL/min;进样方式:分流进样200∶1;辅助加热温度280 ℃;升温程序:初始温度120 ℃保持3 min,以20 ℃/min的速率升至315 ℃,保持5 min,进样量1 μL。
1.6.2 质谱条件
离子源:EI源;离子源温度设置230 ℃,四极杆温度设置150 ℃;电离能量设置70 eV,溶剂延迟8 min;扫描类型:多重反应监测(multiple reaction monitoring,MRM)模式。7种乙酰化糖和糖醇的保留时间、前级离子、子离子和碰撞能量等参数见表1。
2 结果与分析
2.1 质谱条件
MRM模式是利用使用第一级四极杆(MS1)来选择某一质量的前级离子,将其输送至六极杆碰撞反应池进行碎裂,然后通过第二级四极杆(MS2)来监测特征离子碎片。由于MRM模式在第二级四极杆阶段可去除许多化学背景,出现与碎裂离子质量完全相同的同种干扰物机会较少,所以相比单四极杆,三重四极杆质谱定量复杂基质中的低浓度目标物会更加减少化学噪音干扰[17]。在离子源EI模式下对7种挥发性乙酰化糖和糖醇首先进行全扫描一级质谱分析,扫描范围35~660 amu,在所得目标物质谱图中选择质荷比较大且响应强度较高的离子碎片作为前级离子。过低的碰撞能量会导致目标物质无法被电离,而过高的碰撞能量会导致目标物质被轰碎,所以将碰撞能量范围设置5~50 eV(每5eV一个间隔)确定最优碰撞能量,对选定的前级离子峰进行产物离子扫描。前级离子进入二级质谱,将发生断裂或重排等反应产生不同的离子碎片,选取其中响应强度最高的离子作为定量离子,响应强度次之的为定性离子。最后采用MRM模式对目标化合物进行定性定量分析。优化后的质谱分析参数见表1,目标化合物MRM色谱图见图1。
2.2 色谱条件
进样口汽化温度远低于化合物沸点时,化合物连同少量的样品基质会残留在衬管中,可导致后续进样被污染。选取进样口温度分别为260、280、300、320 ℃进行样品检测。对浓度为80 mg/L的混标进行分析,实验结果表明,7七种乙酰化糖和糖的峰面积最大值对应的进样口温度有所不同,但其峰面积均随进样口温度的升高逐渐上升并在300 ℃时基本恒定,故选择的进样口温度为300 ℃。
实验分别考察了弱极性的HP-5MS、中极性DB-1701和强极性DB-23三种毛细管柱对7种糖类化合物的分离效果。结果表明,乙酰化蔗糖在柱温315 ℃可顺利分离,而DB-23与DB-1701的毛细管柱极限温度分别仅达到250 ℃与280 ℃,只可分离其他6种乙酰化糖。HP-5MS毛细管柱极限温度是325 ℃,同时具有超高惰性且柱流失低,可获得较好的峰形和较高的信噪比,因此选取色谱柱HP-5MS为本次实验的分析柱。
乙酰化甘露醇和山梨醇是同分异构体,在SCAN模式下程序升温分离度较差,采用MRM模式分离效果显著改善,与周洪斌等[18]利用降低柱箱升温速率使得乙酰糖分离方法相比,可充分节约检测时间,提高效率。经过调试初始柱温、升温速率、分流模式等参数,7种目标物在15 min内达到基线分离,且检测响应相对较高。图2为MRM模式下7种目标化合物的总离子流色谱图。
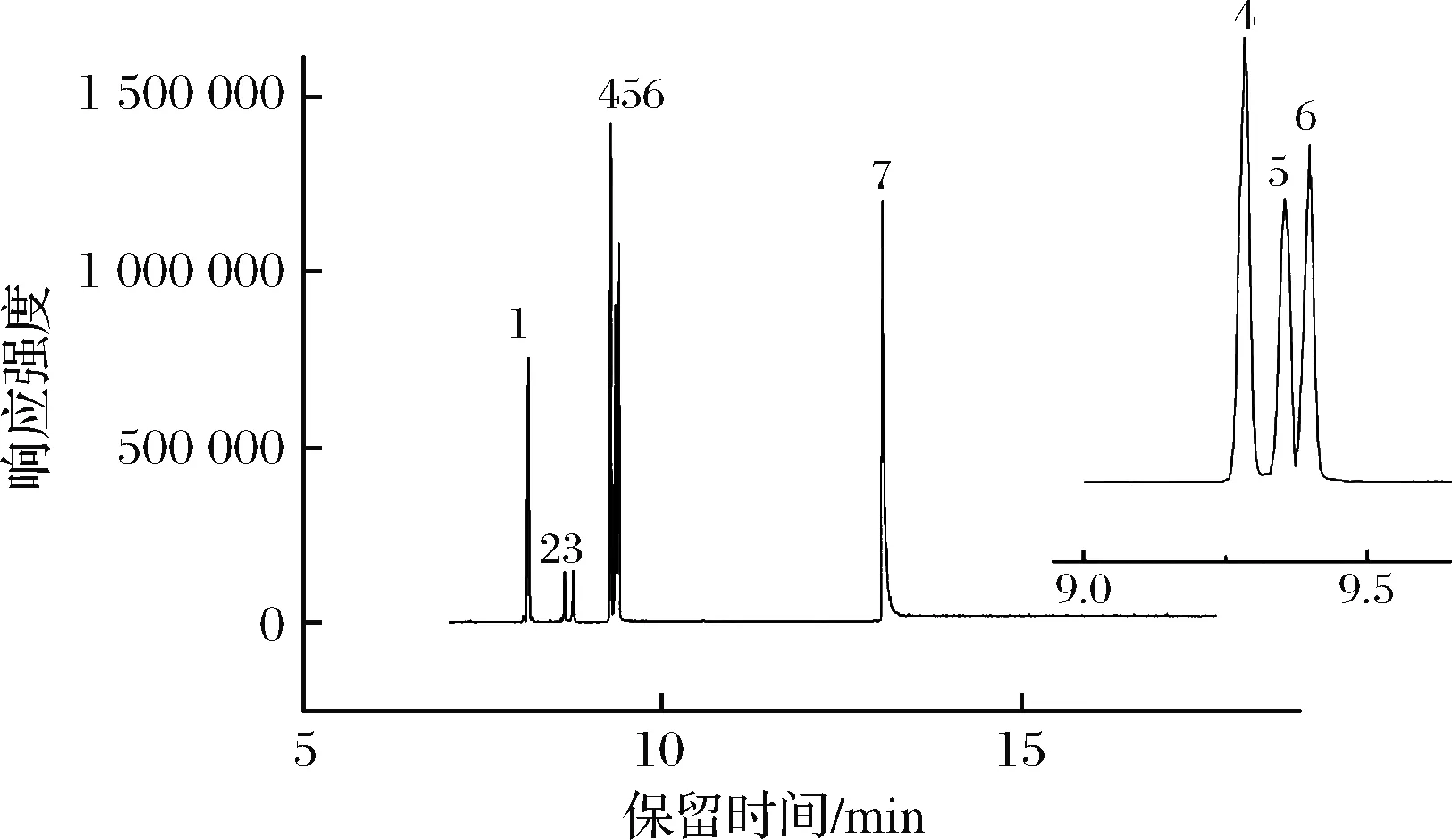
1-侧金盏花醇;2-葡萄糖;3-半乳糖;4-肌醇;5-甘露醇; 6-山梨醇;7-蔗糖图2 七种乙酰化糖和糖醇的总离子色谱图Fig.2 Total ion chromatogram of the 7 aceylated sugars and sugar alcohols
2.3 样品提取方法选择
本次实验采用氯仿-甲醇水溶剂提取法与超声波辅助提取法分别提取枸杞样品中的糖与糖醇,结果如图3所示。采用氯仿-甲醇水溶剂提取法相较超声波辅助提取法所得葡萄糖、肌醇与蔗糖含量略高,但是半乳糖、甘露醇与山梨醇含量略低。2种不同的提取方式均可以提取出糖或糖醇物质且提取效果差异相对较小,考虑到氯仿-甲醇水溶剂提取法操作比较方便、提取液颜色浅,提取用时短,今后实验可选择氯仿-甲醇水溶剂提取法制备样品溶液。

图3 不同提取方法对6种糖和糖醇提取量的影响Fig.3 Effect of different extraction methods on the content of six sugars and sugar alcohols
2.4 MRM方法验证
目标分析物的色谱峰峰型对称且没有基质干扰存在时,可根据保留时间对被分析物进行定性,根据定量离子对的峰面积定量。将1.3项下的标准系列工作液按照优化的分析方法进样测试,为避免前处理过程中出现操作误差,以其他标准品与内标侧金盏花醇的峰面积比为纵坐标,以标准品浓度(mg/L)为横坐标绘制标准曲线,并计算回归方程和相关系数。对标准溶液进行逐级稀释,分别以信噪比(S/N)为10和3时的溶液浓度作为定量限和检出限。结果表明,各化合物线性关系良好,相关系数(R2)均大于0.997,检出限在0.013~0.12 mg/L,定量限在0.045~0.37 mg/L。本方法的线性范围广,灵敏度高。

表2 乙酰化糖和糖醇的工作曲线、相关系数、检出限和定量限Table 2 Regression equations, correlation coefficients,limits of detection and limits of quantification of 6 acetylated sugars and sugar alcohols
选取1组枸杞样品,每份0.2 g,在前处理时分别添加3种不同水平含量的糖和糖醇标准品进行加标回收实验,重复测定6次,如表3显示,每份处理回收率为95.38%~102.6%,相对标准偏差(RSD)为1.28%~3.42%(见表3),该方法精密度良好,达到实际样本检测要求。
2.5 实际样品测量
天然产品药用等级的一个指标是具有药理活性的多糖含量[19]。黑果枸杞属于茄科枸杞属旱生灌木植物,其果实味甘性平,含有大量药理活性的糖类、类胡萝卜素、多酚类等物质,药食两用[20]。芦荟是百合科芦荟属多年生常绿草本植物,具有降血糖,抗肿瘤,抗艾滋以及增强免疫等多种功能,其营养功效主要来自大量的多糖成分。芦荟不同部位含糖量不同,根茎部位的多糖含量较多,叶皮次之,然后是全叶[21]。故本次实验选取枸杞果实与芦荟茎叶部位作为样本,采用氯仿-甲醇水溶剂提取法处理样品,按照已建立的方法测定样品中糖和糖醇的含量,如表4所示。结果表明,黑果枸杞中葡萄糖含量最高,蔗糖含量最低。芦荟中则是半乳糖含量较高,山梨醇含量最低。采用GC-MS/MS多重反应监测模式可以准确地测定植物中糖化合物含量,得到清晰准确的色谱峰。
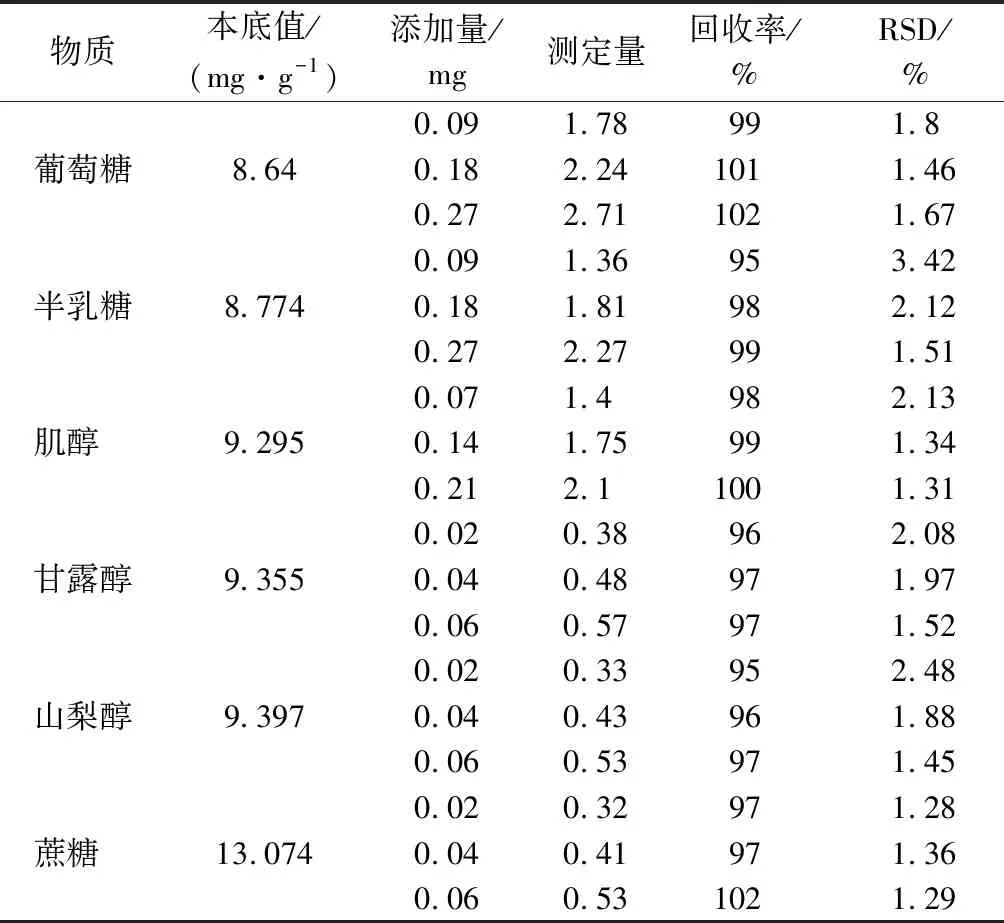
表3 MRM模式下方法的回收率和精密度Table 3 Recoveries and precisions at MRM mode

表4 植物样本中糖和糖醇含量
3 结论
糖类的乙酰化衍生主要经历肟化和乙酰化两步反应。其中肟化反应条件要求较高,反应体系内各组分之间互有协同衍生化反应的影响,需在适宜的温度和反应持续时间下才能顺利向正向反应方向进行[22]。目前对糖类化合物进行乙酰化方法通常使用盐酸羟胺和乙酸酐,1-甲基咪唑或吡啶作为催化剂和溶剂。本次实验中对于样品进行肟化处理时采用吡啶作为溶剂,萃取过程正常,但是采用1-甲基咪唑作为肟化反应溶剂,在萃取中糖溶液不发生分层现象且溶液颜色呈现为黑色,与文献报道[23]存在差异,可能是反应处理时间及其使用量存在不当问题,需要进一步探究。
糖和糖醇乙酰化生成具有挥发性且没有异构峰的糖类衍生物进行气相色谱分离,从而排除了糖类同分异构体的干扰。HP-5毛细管柱在反复使用3个月后,乙酰化糖和糖醇的保留时间位移均在±0.05 min内,考虑仪器误差,认为该方法重现性变化范围符合技术要求。7种乙酰化糖和糖醇通过毛细管色谱柱分离后进入质谱检测,在MRM模式下被分配到不同的通道中,保留时间相对较近的目标物也能完成较好的分离效果。通常多种化合物若在色谱中取得良好的分离效果用时较长,本次实验采用的串联质谱法中多反应监测模式有效地改善了这一不足之处[24]。
本实验通过乙酰衍生化的方法将侧金盏花醇、葡萄糖、半乳糖、肌醇、甘露醇、山梨醇和蔗糖转变为可挥发性物质,结合内标法,建立气相色谱-串联质谱法同时对多种衍生糖化合物进行定性定量分析。实验采用的多重反应监测模式既克服基质效应对响应信号的干扰,又提高实验准确度。该方法样品检测用量少,结果稳定且灵敏度高,可应用于检测植物组织中微量的糖化合物,同时也为检测其他样本中的糖化合物提供更多的选择。
