非综合征型聋儿耳聋基因的检测策略
2021-05-08窦晓宁徐荣华高玲任雪莲张敏敏
窦晓宁,徐荣华,高玲,任雪莲,张敏敏
潍坊市妇幼保健院,山东潍坊261011
遗传性耳聋基因突变是造成儿童听力障碍的主要原因,及时进行耳聋基因检测可以明确大部分听力障碍的原因,揭示遗传性听力障碍的发生发展规律,对于指导医学干预、评估儿童听觉障碍的康复效果及预后有着重要作用。遗传性耳聋基因突变形态复杂多样,有明显的地域特征,固定的筛查基因与位点模板可能存在一定局限性。但基于耳聋基因的广泛性及检测价格成本等因素,在实际工作中无法实现对所有基因及位点进行检测,最有效的方式应是针对本地区的耳聋基因流行情况制定本区域检测的基因与位点模板。本研究对遗传性耳聋基因不同的检测策略进行分析,以期寻找一种科学、合理、经济且检出率高的筛查策略。
1 资料与方法
1.1 临床资料 选取2018年1月—2019年12月经本院听力中心听力学检测及影像学检查(包括耳科常规检查、纯音测听或听性脑干反应、耳声发射、声导抗、颞骨CT 及MR 等)确诊为永久神经性听力损失以及同期潍坊市聋校全部聋生合计非综合征型聋者391 例,男216 例、女175 例,年龄0.2~17(4.4 ±2.8)岁。患儿或监护人在耳鼻喉科医师指导下完成“非综合征型听力障碍者遗传性耳聋基因检测调查问卷”后进行基因检测,调查内容包括一般信息、分娩情况、听障高危因素、家族史、疾病史等。本研究经本院医学伦理委员会审核通过,由患者本人或法定监护人签署《遗传性耳聋基因检测知情同意书》。
1.2 方法 391 例患儿采用随机数字法分成A 组132 例、B 组142 例、C 组117 例,分别进行不同遗传性耳聋热点基因及位点检测。A 组进行遗传性耳聋基因热点基因中的4个基因(GBJ2、SLC26A4、GJB3、12sRNA)21 个位点进行检测。B 组进行遗传性耳聋热点基因6 个基因(GBJ2、SLC26A4、GJB3、12sRNA、POU3F4、GJB6)41 个位点进行检测。C 组进行遗传性耳聋热点基因14 个基因(GBJ2、SLC26A4、GJB3、12sRNA、PRPS1、COCH、DFNAS、DIABLO、DSPP、TECTA、TMC1、GPR98、MYO15A、MYO7A)108 个位点进行检测。检测采用基质辅助激光解吸电离飞行质谱法,所有检出突变病例通过Sanger 一代测序进行验证。
1.3 统计学方法 采用SPSS19.0 统计软件,对各组GBJ2、SLC26A4、GJB3、12sRNA 基因突变频次进行统计,比较各组基因突变率以及GBJ2、SLC26A4、GJB3、12sRNA 基因突变频次。计数资料以例数或百分比表示,比较采用χ2检验。P<0.05 为差异有统计学意义。
2 结果
2.1 三组基因突变情况 三组不同携带类型突变基因及突变位点发生情况见表1。
2.2 基因突变病例Sanger 一代测序验证结果 对3 种检测方式显示存在基因突变的113 例非综合征型耳聋患者进行Sanger 一代测序法验证,其结果与Sanger 一代测序检测均一致,二者的总符合率为100%,Kappa=1。
2.3 三组不同基因突变类型发生例数及总体突变率 各组间总体检出率差异比较无统计学意义(P> 0.05)。见表2。
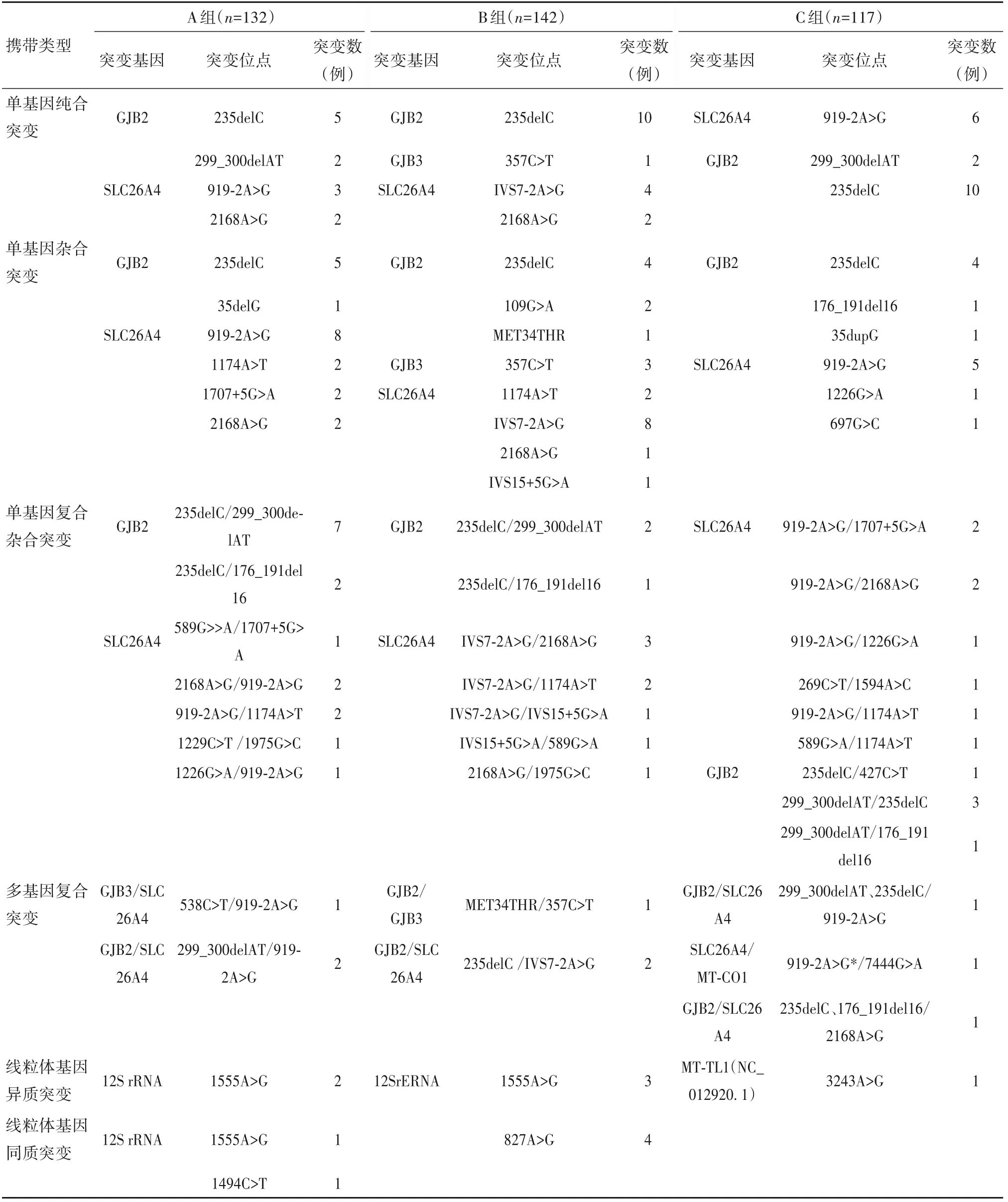
表1 三组基因突变检测结果比较

表2 三组不同基因突变类型发生例数及总体突变率比较
2.4 三组GBJ2、SLC26A4、GJB3、12sRNA 基因突变频次及基因突变率 三种检测方式比较患儿GBJ2、SLC26A4、GJB3、12sRNA 基因突变率差异无统计学意义(P>0.05)。见表3。

表3 三组GBJ2、SLC26A4、GJB3、12sRNA基因突变频次比较
3 讨论
先天性耳聋是最常见的出生缺陷之一,也是最常见的人类感觉系统疾病,发病率为0.1%~0.3%。研究表明遗传因素造成的耳聋占全部耳聋患者的60%~70%,其中约30%为综合征型耳聋,70%为非综合征型耳聋[1]。非综合征型耳聋患者临床上仅出现听觉功能障碍,不伴随其他器官和系统异常,其发病率约为1‰。非综合征型耳聋患者中常染色体隐性遗传常见基因有GJB2、SLC26A4、GJB3、GPR98、TMC1、MYO7A、MYO15A等,占75%~80%;常染色体显性遗传常见基因有GJB3、DSPP、DFNA5、TECTA、DIABLO、COCH 等,占15%~20%;线粒体母系遗传常见基因有MT-RNR1、MT-CO1、MT-TL1、MT-TS1、MT-TH 等,约占1%;X 连锁的遗传性耳聋常见基因有PRPS1等,占2%~5%。目前已知的致病非综合征型耳聋基因175 个,并有新的突变基因不断被发现[2]。耳聋基因检测能够为耳聋患者提供病因分析、干预方法、治疗策略、预防措施、生活方式,并可评估下一代的遗传风险以预防耳聋发生[3-4]。遗传性耳聋基因筛查目前应用的筛查模式多为热点基因筛查,我国遗传性NSHL的最常见致病基因中GJB2、SLC26A4、GJB3、12SrRNA 4 个基因所占的比例较高,上述基因突变携带率分别为3.01%、2.02%、0.37%、0.19%,总的携带率约为6%[5],以上4 个基因突变涵盖遗传非综合征型突变基因的80%左右。常见的突变模式有错义突变、无义突变、移码、插入和缺失等。
GJB2 基因编码的Cx26 表达于耳蜗,位于内耳血管纹、基底膜和螺旋缘,先天性遗传性重度耳聋中大约50%与GJB2 基因突变相关。对遗传性耳聋以往的研究认为,GJB2 基因突变后,K+进入内淋巴液的循环受影响,导致常显或常隐感音神经性耳聋(DFNB1 或DFNA3)。但也有新的研究认为GJB2 的致病机制为柱细胞中微管的减少可能导致螺旋器隧道开放失败,且畸形的指突可能对螺旋器的支持框架产生负面影响[6]。GJB2 复合杂合突变主要影响耳蜗侧壁的间隙连接功能,减弱耳蜗侧壁的杂合耦合,降低耳蜗电位导致耳聋。另有研究认为慢性产前缺氧可导致耳蜗Corti器毛细胞数量减少,Cx26蛋白和mRNA 水平降低,增加基因启动因子区域的甲基化状态,启动因子区超甲基化和Cx26表达下调导致耳聋发生[7]。
SLC26A4 基因编码Pendrin 蛋白质,该蛋白是一种跨膜碘/氯转运蛋白,具有高度疏水性。Pendrin蛋白质功能障碍,可引起Cl-转运障碍或内耳淋巴液流动异常,引起前庭水管扩大和内淋巴管内压升高,内耳毛细胞受损和听神经萎缩,从而导致听力下降,是引起中国人大前庭水管综合征主要原因。目前发现该基因的突变类型有500 多个,且具有显著的种族和地域性[7]。该基因某些突变可导致抗sigma 拮抗剂结构中581 号氨基酸位点上的p. R581M转变,影响SLC26 蛋白稳定性及功能。某些位点的突变可导致细胞质拓扑结构中氨基酸p.Glu704Gln转变,改变Pendrin 蛋白的结构;其构象的随机性增加,减弱了Pendrin蛋白阴离子转运体活性。也有研究认为该基因部分位点突变,存在与GJB2基因突变协同致病的可能性[8]。
GJB3 基因为我国夏家辉院士发现的第一个耳聋相关基因,编码缝隙连接蛋白Cx31,内淋巴钾离子循环障碍,导致内耳毛细胞功能受损,该基因突变可致常染色体显性和隐性非综合征耳聋[7]。起病于青少年期或成人期,通常仅为进行性高频听力受损,少部分患者会有中、重度耳聋。最近有研究认为GJB3 基因编码的Cx31 蛋白与GJB2 基因编码的Cx26 蛋白协同导致耳聋[9]。当GBJ3 基因中c.284C>T 位点与GJB2 基因同时突变时,可能会导致耳聋发生。而有研究认为传统的c.538C>T 位点对听力影响较小。
线粒体基因突变呈母系遗传,依据其在同一个体细胞和组织中的一致程度分为同质性和异质性。线粒体基因1494C>T、1555A>G、7444G>A 突变与氨基糖甙类药物致聋和非综合征性耳聋有关。带有突变的个体对耳毒性药物(氨基糖苷类药物)的敏感性高于正常人,小剂量即可诱发耳聋,通常是高频听力受损,少数患者听力严重受损。线粒体基因突变存在阈值效应,当线粒体基因突变数量达到某个阈值时,可引起相应的临床症状,症状轻重与基因缺陷的严 重 程 度 有 关[10]。有 研 究 认 为tRNAser(UCN)基 因7444G>A 或其他基因位点突变可能为12sRNA 基因1555A>G 突变病理效应的修饰因子,通过影响12sRNA基因1555A>G功能致聋[11]。
本研究将391 例患者随机分为三组,A 组检测4基因21 个位点、B 组检测6 个基因41 个位点、C 组检测14个基因108个位点,此结果与四川、绍兴等地区报告检出率近似[12-13],但与福建、贵州、等地区检出率报告有差异[14-17],此可能与地域性差异以及样本量有关。三组检测的基因因及位点均涵盖了我国大部分地区热点突变基因及位点的80%,而剩余的大约20%已知突变基因数量庞大,即使适量增加检测基因和位点的数量,也不会明显增加其检出率,这可能是三组间检出率无统计学差异的原因之一。本文还显示,4 种常见检测基因GBJ2、SLC26A4、GJB3、12sRNA 突变检出率三组间比较无统计学意义,检测基因和位点增多并不能增加基因突变检测的敏感性及特异性。因此从节约成本角度出发,成本较低的4基因21个位点的检测用于听力障碍耳聋基因筛查即基本能够满足本地需要。
目前,尽管有新的致病基因不断被发现,基因修复也取得一些进展。有研究者利用基因编辑的方法,通过实验动物进行基因编辑获得基因缺陷动物,可以对已有基因缺陷动物进行基因纠正[18]。但是,耳聋基因的致病机理还未完全阐明。无论是耳聋基因的筛查,还是耳聋相关外显子检查等相关诊断,都不能完全排除突变的遗漏。有部分耳聋基因筛查中的基因突变携带者经诊断后确定为复合杂合突变,因此对于筛查出的耳聋基因突变携带耳聋患者应尽可能排查内含子和启动子周围区域内的突变[3]。当然也有部分基因突变患者表现为迟发性耳聋或外显不全,这可能是目前耳聋基因突变的基因型与表型间存在差异性的原因之一。根据目前对耳聋病因的了解,仅靠基因检测来明确病因还不够完善,耳聋的病因检测还应包括病史、孕史及家族史采集、影像学检查、内镜检查以及生化检查等。对于听力正常的突变携带者也不应过度强调耳聋发生的可能性,以免加重突变携带者及家属的心理负担。
