高效液相色谱法测定灵芝二维甲硫氨酸胶囊溶出度及测量不确定度评定
2021-05-07孙煜何虹黄慧朱健萍卢日刚项新华
孙煜,何虹,黄慧,朱健萍,卢日刚,项新华
(1.广西壮族自治区食品药品检验所,南宁 530021; 2.中国食品药品检定研究院,北京 100050)
灵芝二维甲硫氨酸胶囊属于氨基酸维生素类营养药,是以甲硫氨酸、灵芝浸膏、维生素B1、维生素B2为主要原料的胶囊剂,每粒胶囊中含甲硫氨酸25 mg、灵芝浸膏12.5 mg、维生素B12 mg、维生素B21 mg。灵芝二维甲硫氨酸胶囊为口服固体制剂,其现行质量标准为国家食品药品监督管理局国家药品标准WS–10001(HD–1395)–2003,该标准缺少对药品溶出度的检查项目[1]。固体药物的溶出度在很大程度上影响药物的生物等效性和生物利用度,因此需要对固体制剂的溶出度进行严格控制,以保证药物制剂的质量和疗效[2]。为提升并完善本品种的质量标准,更好地控制药品质量,确保用药安全有效,笔者参考有关文献,建立了灵芝二维甲硫氨酸胶囊中三个组分溶出度的测定方法。
溶出度往往由于固体制剂的不均一性、实验系统误差大等因素出现偏差较大的情况,如何科学全面地评价溶出度测量结果,保证仲裁结果的可靠性和权威性,克服传统误差评定方法的局限性,是目前亟待解决的问题。测量不确定度是对测量结果质量的定量表征,其大小反应了测量结果的可信程度以及测量方法的合理性[2–6]。因此,当溶出度结果出现边缘值而涉及符合性判断时,测量不确定度的评定就显得格外重要。
笔者结合灵芝二维甲硫氨酸胶囊溶出度测定方法及数据结果,首次在药品检测领域,依据标准RB/T 141–2018 《化学检测领域测量不确定度评定利用质量控制和方法确认数据评定不确定度》[7],采用Top-down 不确定度评定法对其各组分测量不确定度进行评定。该方法是一种自上而下的不确定度评定法,是在期间精密度测量条件下,将期间精密度和偏倚的累积效应合并,视为不确定度的估计量。Top-down 评定方法简便,操作性强,可以最大限度地避免影响不确定度分量的不确定性、交互效应无法估计以及非正态繁琐的数学运算等问题[5–9]。
1 实验部分
1.1 主要仪器与试剂
电子分析天平:XS205DU 型,感量为0.01 mg,德国赛多利斯仪器有限公司。
高效液相色谱(HPLC)仪:(1)Thermo Ultimate 3000 型,美国赛默飞世尔公司;(2)Waters2695–2487/996 型,美国沃特世公司。
溶出度及自动取样系统:EDT–14Lx 型,印度Electrolab 公司。
甲硫氨酸对照品:批号为140684–201102,含量为100%,中国食品药品检定研究院。
维生素B1对照品:批号为100390–201505,含量为97.0%,中国食品药品检定研究院。
维生素B2对照品:批号为100369–201504,含量为98.2%,中国食品药品检定研究院。
灵芝二维甲硫氨酸胶囊样品:某企业提供。
甲醇、甲酸:均为色谱纯。
甲酸铵、盐酸:均为分析纯。
1.2 色谱条件
色谱柱:Waters Atlantis T3 或Thermo syncronis aQ 柱(250 mm×4.6 mm,5 μm);检测器:二极管阵列检测器(DAD);流动相:A 为水–1.0 mol/L甲酸铵–甲酸(990∶10∶1),B 为甲醇–1.0 mol/L甲酸铵–甲酸(990∶10∶1);梯度洗脱(程序见表1);流动相流量:1.0 mL/min;检测波长:甲硫氨酸为212 nm,维生素B1与维生素B2为260 nm;进样体积:20 μL。
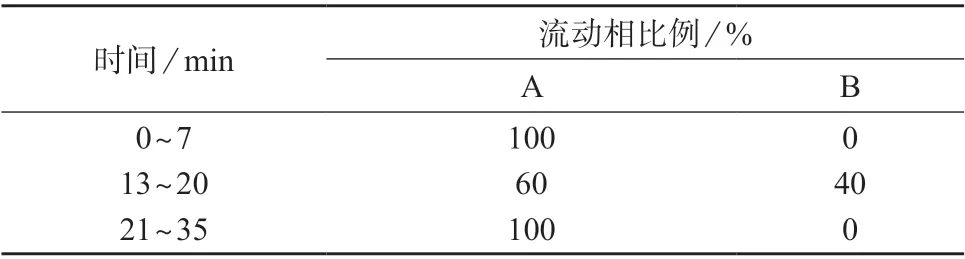
表1 线性梯度洗脱程序
参考文献[10–22]建立溶出度检查的高效液相色谱方法。
1.3 对照品溶液配制
甲硫氨酸对照品储备液:1 mg/mL,取甲硫氨酸对照品适量,用0.1 mol/L 盐酸溶液溶解并稀释制得。
维生素B1对照品储备液:0.05 mg/mL,取维生素B1对照品适量,用0.1 mol/L 盐酸溶液溶解并稀释制得。
维生素B2对照品储备液:0.01 mg/mL,取维生素B2对照品约10 mg,置于100 mL 容量瓶中,加0.1 mol/L 盐酸溶液适量,置于65℃水浴中振摇20 min,再超声5 min 使之溶解,用溶出介质稀释制得。
混合对照溶液:分别取上述3 种对照品储备液0.75、1.0、2.5 mL 于同一25 mL 容量瓶中,用0.1 mol/L 盐酸溶液稀释至标线。其中甲硫氨酸质量浓度约为0.03 mg/mL,维生素B1质量浓度约为2 μg/mL,维生素B2质量浓度约为1 μg/mL。
1.4 样品处理
取灵芝二维甲硫氨酸胶囊样品,按《中国药典》2015 年版四部通则0931《溶出度与释放度测定法第二法 桨法》,以900 mL 0.1 mol/L 盐酸溶液为溶出介质,搅拌器转速为75 r/min,搅拌30 min 后取溶出液10 mL,用0.45 μm 滤膜滤过,取续滤液作为样品溶液。
2 结果与讨论
2.1 溶出介质的选择
分别考察了0.1 mol/L 盐酸溶液、pH 4.5 的乙酸盐缓冲液、pH 6.8 的磷酸盐缓冲液及水4 种溶出介质,其中,pH 4.5 的乙酸盐缓冲溶液在212 nm 波长处甲硫氨酸峰保留时间位置有明显溶剂峰,对甲硫氨酸峰干扰明显;维生素B2在水中溶解度较低,其对照品不易制备成水溶液,且维生素B2进入体内消化系统后主要在小肠上段被吸收,快速释药有利于增加其生物利用度[15]。综合上述几点,最终选择接近胃酸中溶解环境的0.1 mol/L 盐酸溶液作为溶出介质。
2.2 搅拌转速及溶出时间的选择
分别考察了灵芝二维甲硫氨酸胶囊样品在0.1 mol/L 盐酸溶液作为溶出介质条件下,在50 r/min 及75 r/min 转速下120 min 内的溶出曲线,结果显示,75 r/min 转速条件比50 r/min 转速条件更具有区分力,3 个组分30 min 溶出度均能达到75 %。因此最终选择转速为75 r/min,溶出时间为30 min。
2.3 流动相的选择
笔者曾采用甲醇–0.07 mol/L 庚烷磺酸钠溶液(加14 mL 三乙胺并加水至1 000 mL,用磷酸调节pH 至3.5)作为流动相[10],梯度洗脱,但色谱图中会因流动相梯度变化而出现一个峰,该峰大小与目标成分色谱峰接近,容易造成分析干扰;并且该流动相组成为离子对试剂,对色谱柱的损耗较大。参考文献[5–14]选择对流动相组成和梯度洗脱程序进行了优化,最终流动相组成与梯度洗脱程序见1.2。
2.4 检测波长的选择
取对照品溶液,按1.2 色谱条件进样,用DAD检测器在200~400 nm 波长范围进行紫外吸收光谱扫描。结果显示,在该色谱条件下,甲硫氨酸最大吸收波长约为212 nm,维生素B1最大吸收波长为247 nm,维生素B2最大吸收波长为224 nm 及269 nm。为兼顾3 组分的测定灵敏度,选择检测波长为212 nm 测定甲硫氨酸,检测波长为260 nm 同时测定维生素B1、维生素B2。取对照品溶液及样品溶液,按照1.2 色谱条件进行测定,记录色谱峰的保留时间和峰面积,按外标法以峰面积计算各组分溶出量。图1、图2 分别为对照品、样品溶出度试验色谱图。
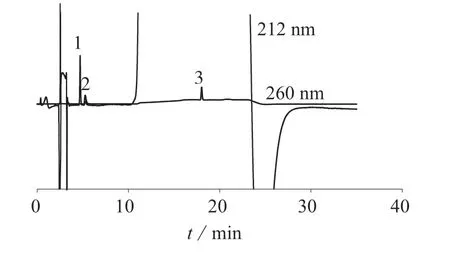
图1 混合对照品溶液色谱图
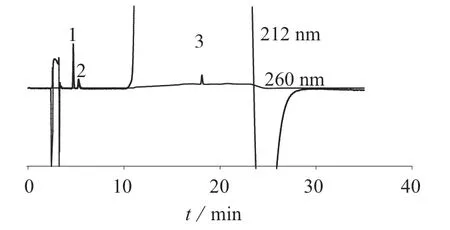
图2 样品溶液色谱图
2.5 线性关系
分别精密称取甲硫氨酸对照品、维生素B1对照品、维生素B2对照品适量,用0.1 mol/L 盐酸溶液制成系列浓度的标准工作溶液(见表2)。按1.2 色谱条件进样分析,记录色谱峰面积。分别以甲硫氨酸、维生素B1和维生素B2的色谱峰面积为纵坐标(Y),溶液的质量浓度为横坐标(X)进行线性回归,得回归方程分别为:Y=110.4X –0.013 59,Y=0.466 8X–0.048 79,Y=0.892 4X–0.096 52,相 关 系数分别为1.000 0,1.000 0,0.999 9。结果表明,甲硫氨 酸 在0.010 38~0.051 92 mg/mL、维 生 素B1在1.113~5.563 μg/mL、维生素B2在0.600 0~3.000 μg/mL 范围内线性关系良好。
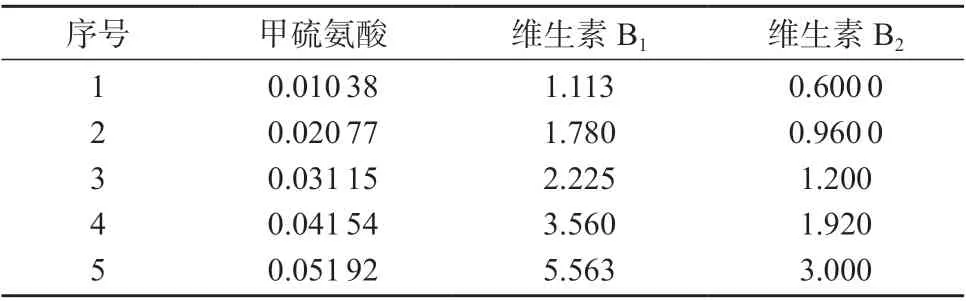
表2 系列标准工作溶液的质量浓度 μg/mL
2.6 稳定性试验
取中间浓度的标准工作溶液,按照1.2 色谱条件连续进样分析,持续32 h,结果显示,溶液在32 h内稳定,甲硫氨酸、维生素B1和维生素B2色谱峰面积的相对标准偏差分别为1.3 %、0.47 %和0.32 %(n=17)。
2.7 加标回收试验
按处方比例,配制混合空白样品,称取空白样品约0.033 g 共9 份,分别置于9 只100 mL 容量瓶中,分别按处方含量的80%、100%、120%加入对照样品,即加入1.0 mg/mL 甲硫氨酸对照品储备液2.0、3.0、4.0 mL;0.2 mg/mL 维生素B1对照品储备液0.8、1.0、1.2 mL;0.1 mg/mL 维生素B2对照品储备液0.8、1.0、1.2 mL。用0.1 mol/L 盐酸溶液稀释并定容,摇匀,按照1.2 色谱条件进样分析,以色谱峰面积外标法计算回收率。
结果表明甲硫氨酸、维生素B1和维生素B2的回收率良好,平均回收率分别为101.5%、99.4%、99.5%,测定结果的相对标准偏差分别为0.8%、0.4%和0.9%(n=9)。
3 测量不确定度评定
采用Top-down 法进行测量不确定度的评定。根据RB/T 141–2018 《化学检测领域测量不确定度评定 利用质量控制和方法确认数据评定不确定度》[7],将测量不确定度来源分为两方面,即随机效应和系统效应。随机效应主要通过期间精密度(uR’)来量化,系统效应通过实验室偏倚(ub)来量化。其中,按照RB/T 141–2018 要求,同时结合本实验室标准溶液样品特点,期间精密度uR’按照标准溶液样品的变异(中间精密度数据结果)及实际样品不同水平之间的变异(重复性数据结果)来评定;实验室偏倚ub按照多水平加标回收试验结果的偏倚来评定。
3.1 数学模型及测量不确定度来源
3.1.1 数学模型
根据标准RB/T 141–2018,各组分溶出量的不确定度按式(1)计算:
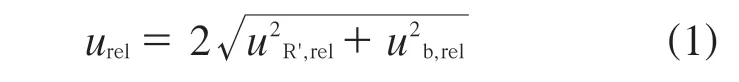
式中:uR’,rel——期间精密度不确定度分量;
ub,rel——偏倚不确定度分量。
3.1.2 测量不确定度来源
根据式(1)确定测量不确定度的来源有期间精密度、实验室偏倚两项。
期间精密度引入的不确定度分量:来自于方法学验证过程中标准溶液中间精密度试验数据的偏差以及实际样品多次重复测定结果的偏差引入的不确定度。按照式(2)计算:
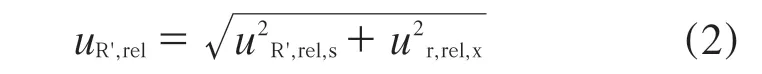
式中:uR’,rel,s——标准溶液中间精密度不确定度分量;
ur,rel,x——样品重复性不确定度分量。
偏倚不确定度分量:来自于方法学验证过程中加标回收试验引入的不确定度。
3.2 测量不确定度评定
3.2.1 标准溶液测定结果
本样品为高溶解性药物制剂,依据《普通口服固体制剂溶出度试验技术指导原则》[23]的要求,采用单一浓度检测各组分溶出量。
选取同水平标准溶液(即混合对照溶液)中间精密度17 个试验数据进行Anderson-Darling 检验(即安德森–达令检验,以下简称AD),各组分的AD 检验结果均小于1.0,说明数据在99%概率下接受测量系统的正态性与独立性假设。数据的相对标准偏差则视为ur’,rel,s。标准溶液测定结果见表3。
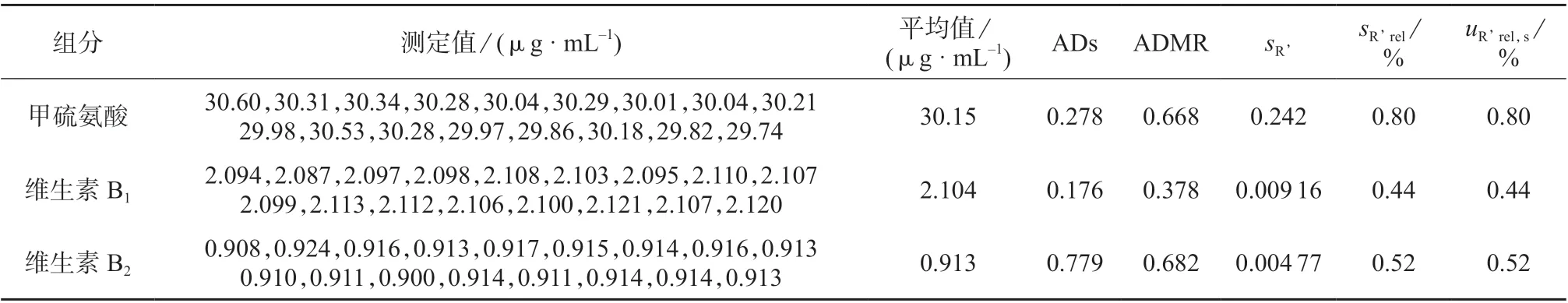
表3 标准溶液各组分质量浓度测定结果
3.2.2 样品测定结果
随机选取样品重复试验数据,对其中6 组数据进行AD 检验,各组分测量数据均通过AD 正态及独立性检验,其不确定度分量:
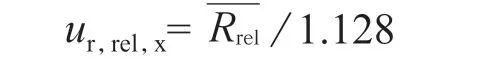
样品中甲硫氨酸、维生素B1、维生素B2溶出度测定数据分别见表4、表5、表6。
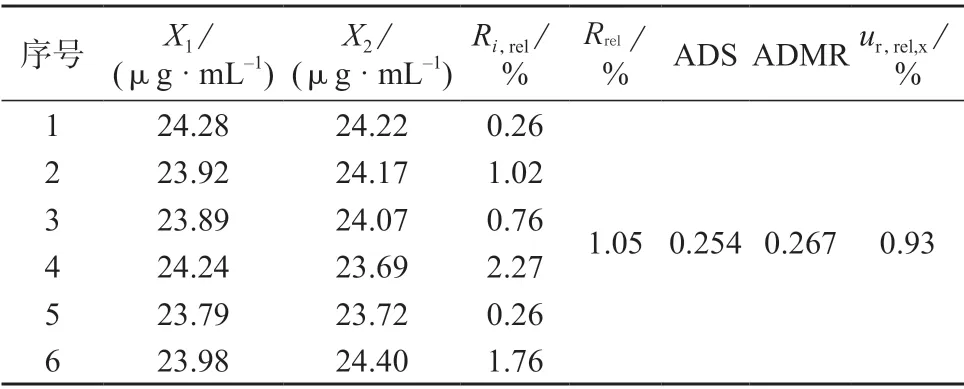
表4 样品中甲硫氨酸溶出度测定数据
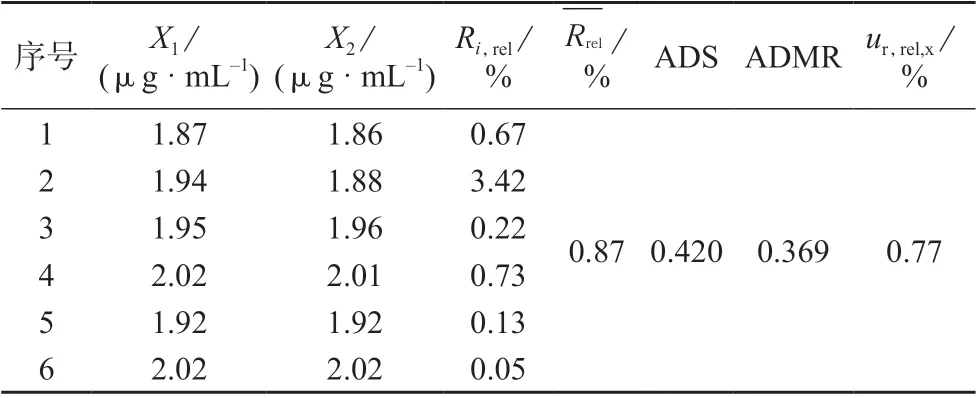
表5 样品中维生素B1 溶出度测定数据
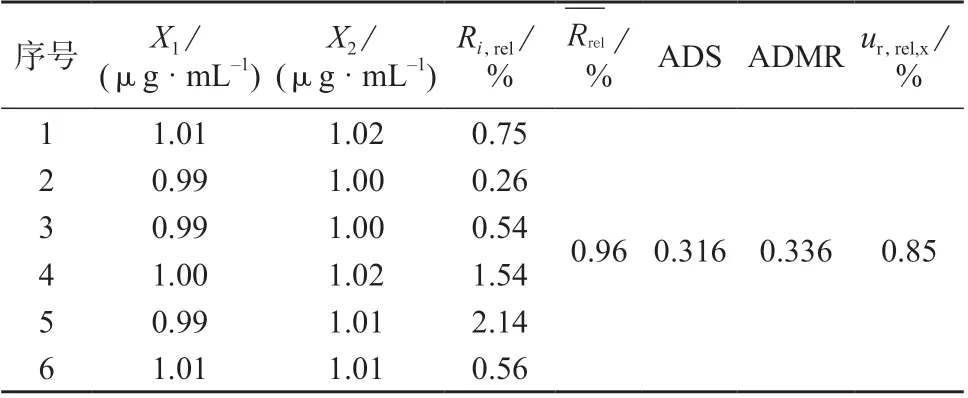
表6 样品中维生素B2 溶出度测定数据
3.2.3 期间精密度不确定度分量
利用标准溶液及样品分析得到期间精密度,按式(2)计算不确定度分量uR’,rel,得到甲硫氨酸、维生素B1、维生素B2的期间精密度相对不确定度分量uR’,rel分别为1.23 %、0.89 %、1.00 %。
3.2.4 偏倚不确定度分量
根据加标回收试验3 个加标浓度水平(120%、100%、80%)下的回收率数据进行偏倚不确定度的评定,得到各组分偏倚不确定度分量ub,rel=brms,rel[7]。加标回收率数据及计算结果见表7~表9。
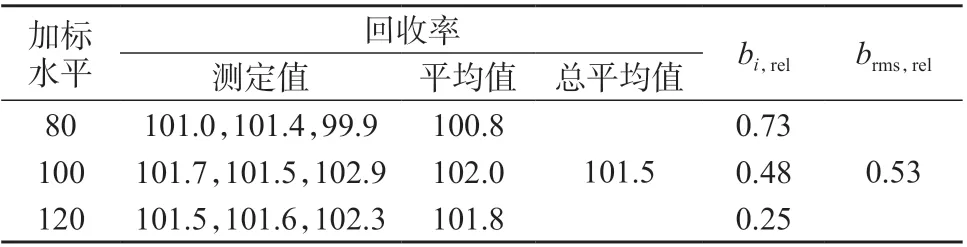
表7 甲硫氨酸加标回收试验结果 %
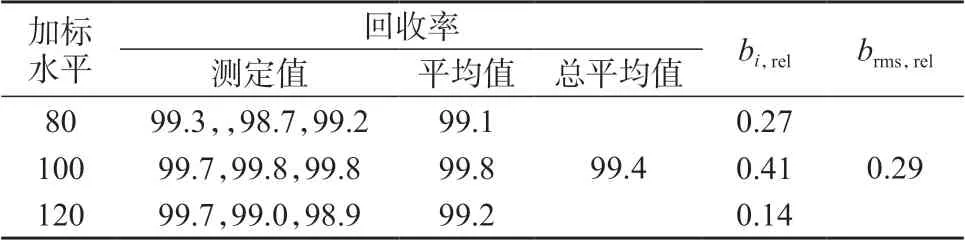
表8 维生素B1 加标回收试验结果 %
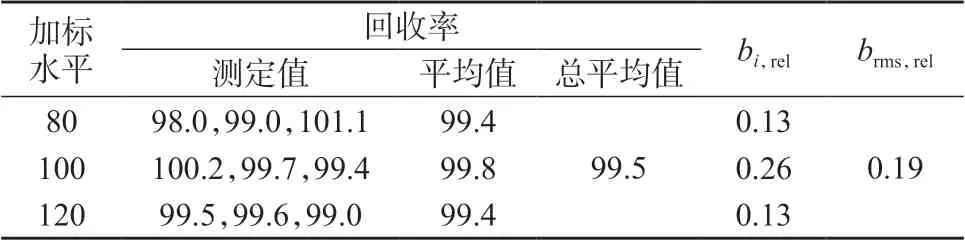
表9 维生素B2 加标回收试验结果 %
3.2.5 扩展不确定度
将期间精密度不确定度分量及偏倚不确定度分量进行合成,得到合成不确定度,包含因子k=2,在95%概率下相对扩展不确定度按式(1)计算,得到甲硫氨酸、维生素B1、维生素B2的溶出度结果相对扩展不确定度分别为2.7%、1.9%、2.0%。测得样品中甲硫氨酸、维生素B1、维生素B2的溶出度分别为92%、90%、84%,包含概率95%下的溶出度结果分别表示为(92±2.7)%、(90±1.9)%、(84±2.0)%。
4 结语
建立了灵芝二维甲硫氨酸胶囊溶出度的检查方法,完善了现行质量标准。采用HPLC 法同时测定甲硫氨酸、维生素B1和B2的溶出量,操作简便,数据科学、准确。首次采用Top-down 不确定度评定法对化学药品检测进行不确定度评定,拓展了该方法在药品检测领域中的应用。
