苝酰胺醇酯的光物理和电化学性质
2021-05-06刘丽敏石建军张艳红马祥梅
刘丽敏 石建军 张艳红 马祥梅
(安徽理工大学化学工程学院 淮南232001)
苝-3,4,9,10-四羧酸二酰亚胺(通常称为苝二酰亚胺,PDIS)及其衍生物是一类杰出的有机化合物,广泛应用于有机太阳能电池[1-3]、光导体、场效应晶体管[4-5]、传感器[6-7]、有机发光二极管[8]、光敏剂[9]、光开关、生物荧光探针等[10-12]方面。PDIS 的分子结构包括一个刚性芳香族核(苝)和两个亚胺环。富电子的苝核与强吸电子的酰亚胺基团之间的π聚集扩展导致其电子吸收带发生红移,移至约520 nm 附近,这是电子震动导致的特殊进程。因此,这类化合物表现出高的化学、热、电化学和光化学稳定性以及高的荧光量子产率。从1913年第一次被发现至今[13],研究者们关注的重点一直是各种PDIS结构的合成,结构-性能关系,PDIS 在光子学和有机电子中的应用,关于它们的超分子结构,包括氢键相互作用和有机金属结构。
用于修饰PDIS 结构的两种主要途径包括:在“bay”区域(1,6,7,12-位)和在“peri”位置(酰亚胺)引入不同取代基[14-15]。“bay”区域功能化可以调节PDIS 染料的光电性质以及通过改变苝核的平面结构有效改善其溶解性[16]。酰亚胺位置上的取代基可以调节相应苝酰亚胺的聚集性、溶解性和电子传输率,同时对其吸收和荧光等光电性质也有一定影响[17-18]。这是由于分子前沿轨道能量节点产生的HOMO/LUMO 能级仅对酰亚胺上的N原子有效[19]。因此,合成新的N原子取代PDIS,研究取代基对其光学、光物理和电化学性能的影响以及对PDIS 光电性能调节规律仍然具有重要意义。
众所周知,酰亚胺位置取代基对苝系染料的共轭体系没有影响或影响很小[20-21]。然而,假设酰亚胺位置取代基与苝系染料的共轭体系没有关系也是不合理的。本文研究了烷基和羧酸酯对苝酰亚胺荧光和电化学性质的影响,测定并比较了六种酰胺酯(结构见图1和表1)的吸收和荧光性能,以及电致发光性能,并探讨了烷基链长短对这些染料电化学和荧光性能的影响,为揭示端位置取代对苝酰亚胺类衍生物光物理和电化学性质的影响机理,以及为满足各种应用需要而调节的光电性质提供了一定的指导。
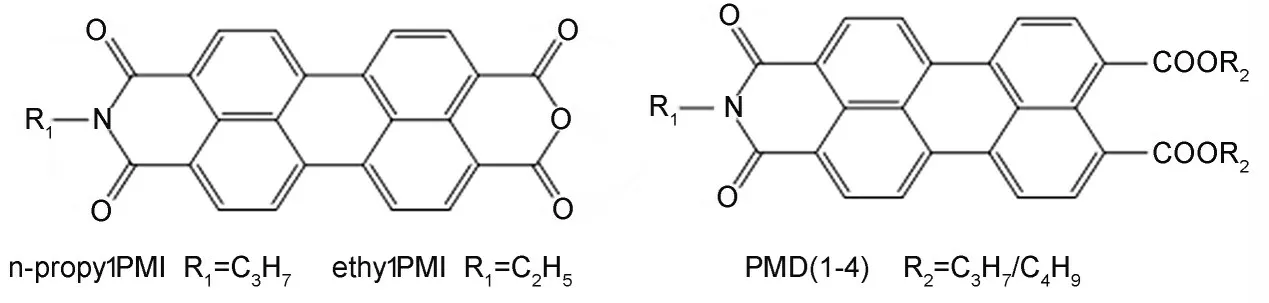
图1 化合物结构式Fig.1 Chemical structure of compounds
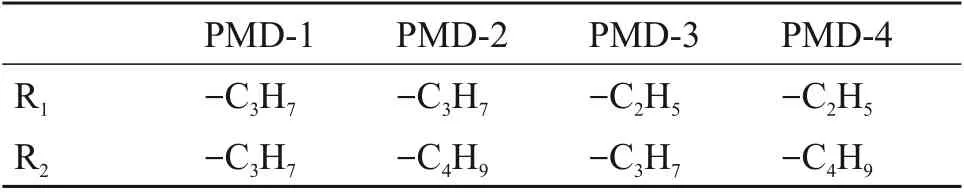
表1 PMD(1~4)取代基Table 1 Substituent groups of PMD(1~4)
1 材料与方法
1.1 试剂
所用试剂均购自国药集团化学试剂有限公司,均为分析纯,所用水为去离子水。苝酰亚胺醇酯按照Nagao 等[22]的方法合成,采用重结晶和柱色谱进行分离提纯,经硅胶层析板点样检测,UVVis 谱及IR 谱与文献[23]报道相符(纯度约为99.7%)。DMF 等溶剂均为重新蒸馏过的分析纯溶剂。
1.2 荧光和吸收光谱溶液配制
称量化合物ethylPMI、n-propylPMI 和PMD(1~4)溶解至10 mL氯仿中得到0.1 mmol/L化合物溶液A,再取少量溶液A 稀释至波长480 nm 处吸光度为0.100 的溶液B,溶液B 用于测量吸收和荧光光谱、荧光量子产率。
1.3 仪器与测试方法
荧光光谱用F-4600 荧光光谱仪测定,为天津科美有限公司产品,激发光源为450 W氙灯,激发波长480 nm。UV-Vis 吸收光谱用UV-2550 紫外-可见分光光度计测定,光程10 mm。
荧光量子产率Φf在氯仿中测量,用式(1)计算。参比苝酐在DMF 中的荧光量子产率为Φ0=0.99[24]。

式中:F 表示荧光强度(a.u.);A 表示激发波长处的吸光度;n为折光率;S表示样品;0表示参比。
电化学性质通过循环伏安法(CV)测得,工作电极为三氧化二铝打磨后经PMD(1~4)修饰的玻碳电极(三氧化二铝粒径0.05 μm,6 μL 1.01×10-6mol/L 的PMD(1~4)滴涂在电极表面),参比电极为饱和甘汞电极,对电极为铂电极,电解质为溶解在PBS 缓冲液中的0.1 mol/L 的K2S2O8;室温扫描,扫描范围为-1.6~1.0 V,扫描速率为0.1 V/s。电致化学发光(ECL)用MPI-E 电化学分析仪(西安迈瑞分析仪器有限公司产品)测定。把化合物溶解在氯仿中,滴在工作电极上,烘干制得薄膜,测量时间1 000 s,采样速率10 T/s,放大级数3,光电倍增管高压800 V,其他测定参数同CV法。
2 结果与讨论
2.1 光学性质
图2 是化合物ethylPMI、n-propylPMI、PMD(1~4)在氯仿中的紫外-可见吸收光谱(其浓度为1.01×10-6~6.79×10-6mol/L),呈现了π-π*跃迁的特征强吸收,其S0→S1的电子跃迁分别位于518 nm、520 nm、525 nm、525 nm、524 nm 和521 nm(最大吸收峰)处,与苝二酰亚胺(在氯仿中的最大吸收波长位于504 nm处[21])相比,化合物的最大吸收峰明显红移,这是苝核和取代基之间的电子相互作用所致。烷基酯化的苝酰亚胺与苝酰亚胺单酐相比红移7 nm。这是因为酸酐键的酯化降低了它的拉电子能力。除了较低能量的S0→S1电子吸收带以外,还观察到化合物属于S0→S2电子跃迁的吸收带,分别位于453 nm、456 nm、454 nm、458 nm、458 nm 和451 nm 处,与较长波段的吸收相比这些波段的吸收相对较弱。对于n-propylPMI 来说,羧基烷基化对其最大吸收波长几乎没有影响,而其S0→S2电子吸收带却红移4 nm。对于ethylPMI 来说,其吸收光谱随着碳原子数的增多发生蓝移,两个吸收带分别蓝移3 nm 和7 nm。这与理论上随着碳原子数的增多烷基给电子能力增大相反,且PMD-4具有显著的谱带变窄。这可能是因为ethylPMI在氯仿中更容易形成H 聚集体,而n-propylPMI 丙基取代基空间位阻相对较大,阻碍了H 聚集体的形成,使其更容易形成J聚集体[25-26]。计算所得的对应最大吸收点的摩尔消光系数以及其他相关的光物理数据如表2所示。

图2 化合物n-propylPMI、ethylPMI(a)和PMD(1~4)(b)在氯仿中的紫外-可见吸收光谱Ccompounds=1.01×10-6~6.79×10-6 mol/LFig.2 UV-Vis spectra for compounds of n-propylPMI,ethylPMI(a)and PMD(1~4)(b)in chloroform
对于有机化合物,其荧光与化合物的结构有着密切的关联。一般的情况下,分子的荧光与分子的共轭体系、刚性的平面结构及电子跃迁类型有关。强的荧光物质通常有较大的共轭π键体系及刚性平面结构,这是因为任何能引起分子振动或转动增加(可导致激发态分子的能量损失)的结构或基团都能使分子产生荧光淬灭[21]。
图3 为化合物ethylPMI、n-propylPMI、PMD(1~4)在氯仿中的荧光发射光谱。与吸收光谱相似,与n-propylPMI 和ethylPMI 相比化合物的最大发射波长都有一定的红移,约2~3 nm。这是因为酸酐键酯化降低了其拉电子能力。对于npropylPMI 来说,随着酯化碳原子数的增加峰位置有一定红移;而ethylPMI 却与n-propylPMI 相反,随着酯化碳原子数的增加峰位置蓝移4 nm。这与人们通常认为的随着端位取代基给电子能力增加峰位置发生红移相反。
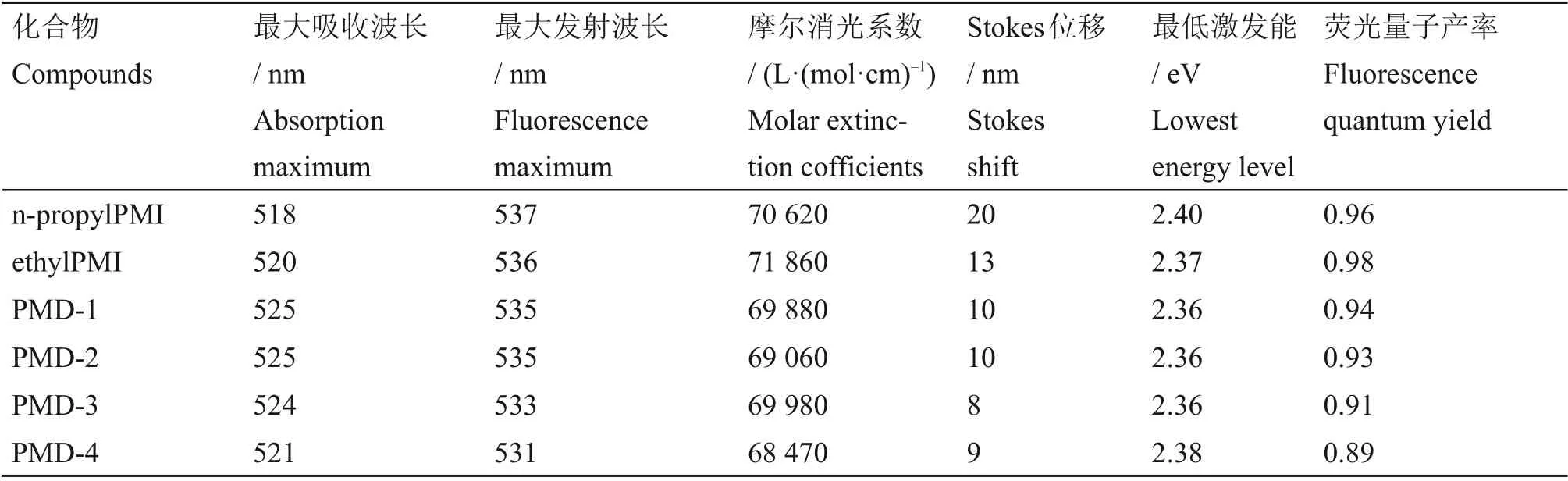
表2 化合物在氯仿中的光物理数据Table 2 Photophysical data of compounds in chloroform
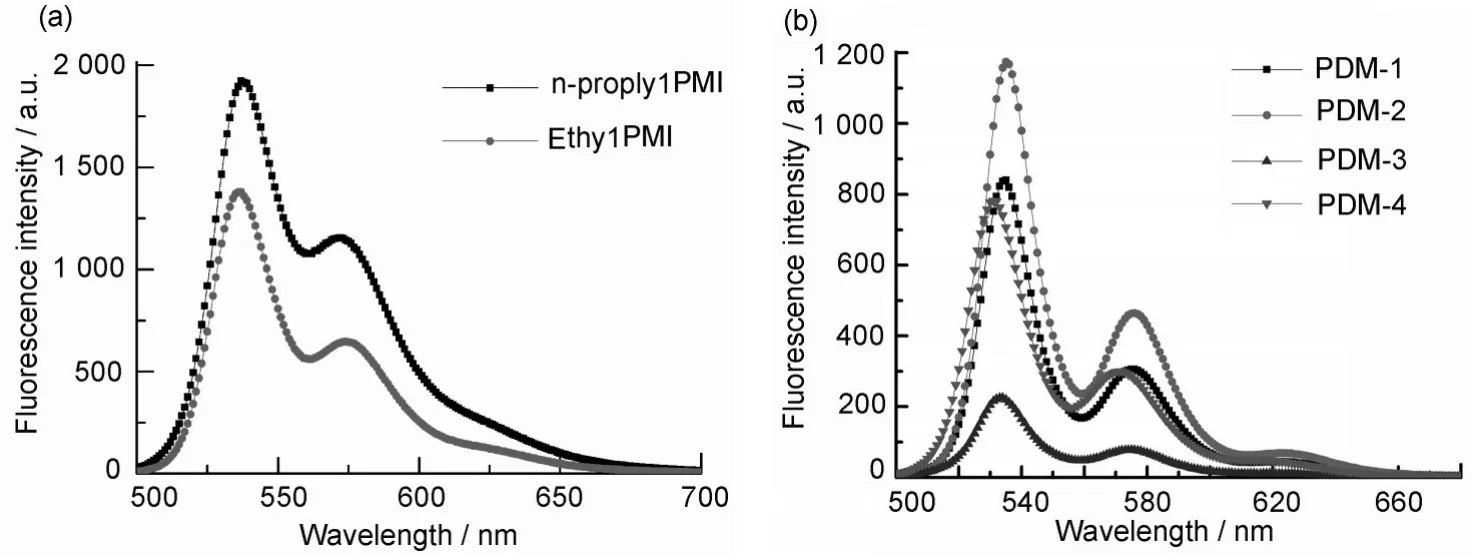
图3 n-propylPMI、ethylPMI(a)和PMD(1~4)(b)在氯仿中的荧光光谱,激发波长480 nmFig.3 Fluorescence spectra of n-propylPMI,ethylPMI(a)and PMD(1~4)(b)in chloroform,λex=480 nm
测定的化合物ethylPMI、n-propylPMI 和PMD(1~4)的荧光量子产率(Φf)如表2所示。对于这6种化合物而言,它们的荧光量子产率随着酸酐键酯化及酯化碳链的增长而略有降低,这是因为在端位引入给电子取代基使其激发态发生了电荷转移,从而造成了荧光量子产率一定程度的减小。对于其Stokes 位移和最低激发能(Es)却影响不大,表明酸酐键酯化没有改变π-共轭体系基态和激发态的几何构型。
2.2 电化学性质
图4 是化合物PMD(1~4)的循环伏安曲线,扫描电压从-1.6~1.0 V。PMD-2和PMD-4只有还原峰(其电位分别为-0.97 V 和-0.89 V)而无氧化峰,每次测量扫描10 次,峰值、强度和位置变化很小。然而,PMD-1 和PMD-3 的循环伏安曲线中没有出现还原峰,无论是从0 到负再到0,还是施加正电位都没有出现还原峰。由此可知,苝系衍生物酸酐酯化的碳链越长化合物越容易被氧化。

图4 化合物PMD(1~4)的循环伏安曲线Fig.4 Cyclic voltammograms of PMD(1~4)
ECL 测量表明:化合物PMD(1~4)分别在-1.07 V、-1.12 V、-1.10 V、-1.14 V 电位处开始发光(如图5 所示)。其中PMD-2 的发光强度最大,PMD-4 次之,且化合物PMD-1、PMD-3 在200 s 之后稳定在1 200 的光强上,化合物PMD-2在475 s 后稳定在7 000 的光强上,化合物PMD-4在250 s 后稳定在2 400 的光强上(如图6~7所示)。
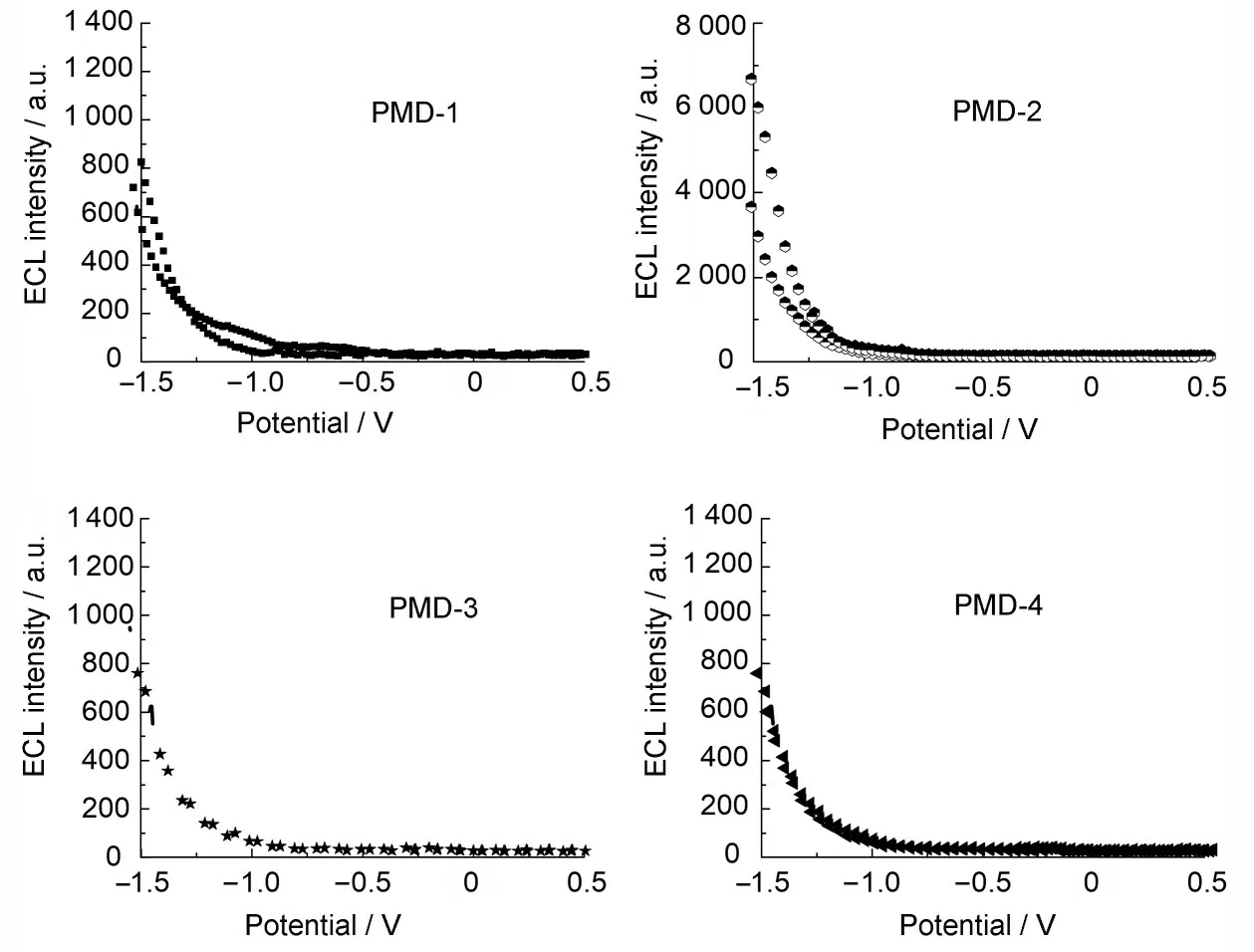
图5 化合物PMD(1~4)的发光强度-电位曲线Fig.5 Luminescence intensity-potential curves of PMD(1~4)
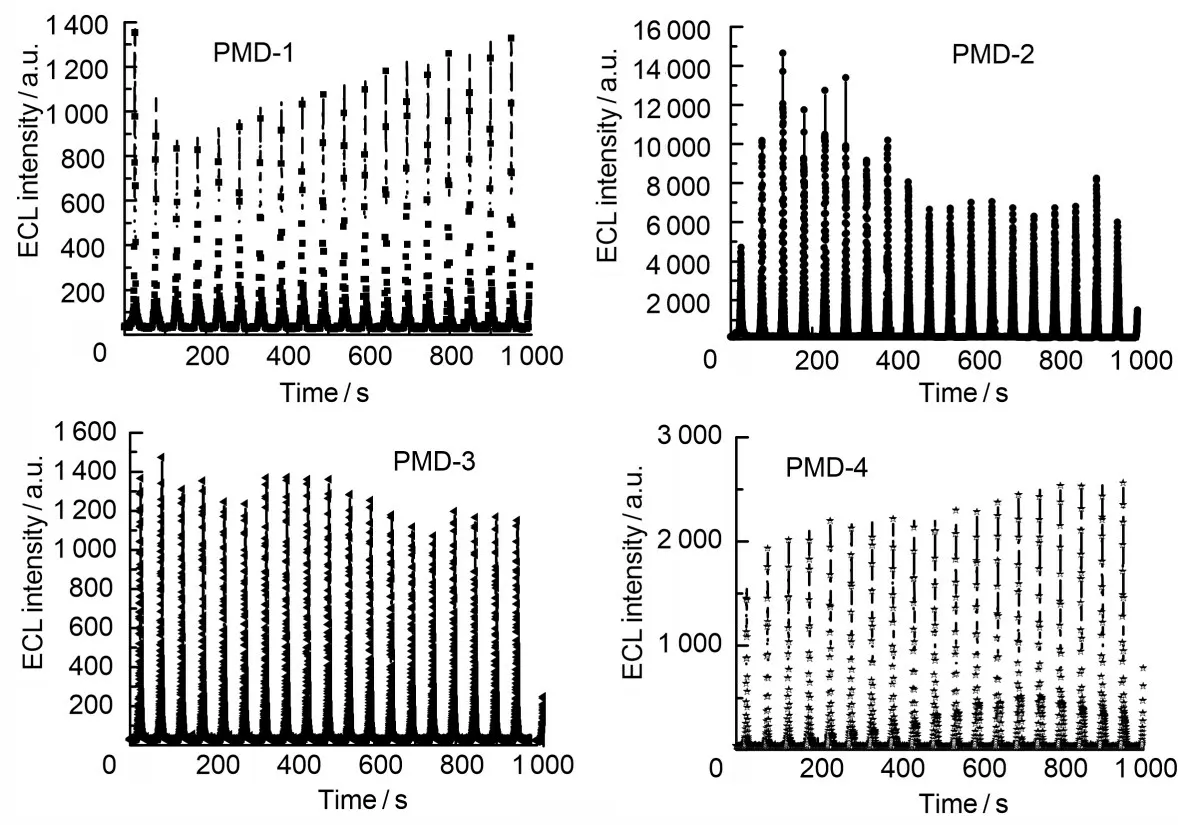
图6 化合物PMD(1~4)ECL稳定性Fig.6 Stability of ECL for PMD(1~4)
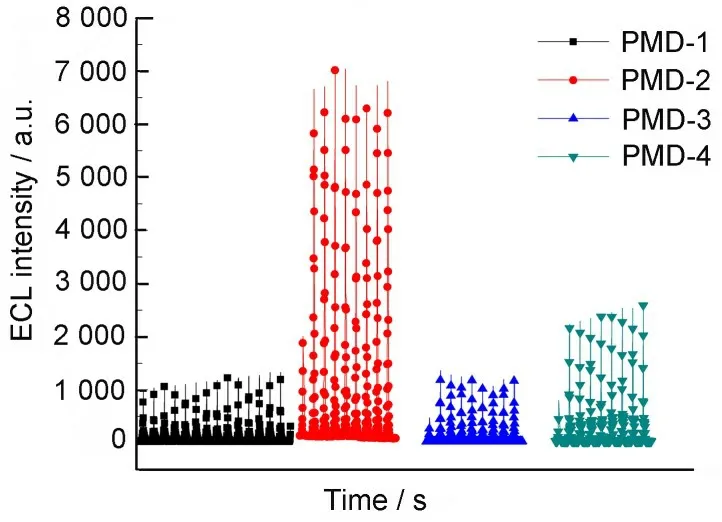
图7化合物PMD(1~4)的ECL强度对比Fig.7 Comparison of ECL intensity for PMD(1~4)
3 结论
通过UV-Vis 和荧光光谱对化合物npropylPMI、ethylPMI 和PMD(1~4)的光学和光物理性质进行了表征。最大摩尔消光系数分别为70 620 L/(mol·cm)、71 860 L/(mol·cm)、69 880 L/(mol·cm)、69 060 L/(mol·cm)、69 980 L/(mol·cm)和68 470 L/(mol·cm),在可见光区400~600 nm 处具有高光子吸收能力,使得这些化合物非常适合作为光活性材料的衍生物。除了较低能级的S0→S1电子吸收带以外,还观察到化合物属于S0→S2电子跃迁的吸收带,分别位于453 nm、456 nm、454 nm、458 nm、458 nm 和451 nm 处。另外,酸酐键酯化增加了化合物的溶解度,化合物PMD(1~4)易溶于低极性溶剂(如氯仿),且它们的荧光量子产率随着酸酐键酯化及酯化碳链的增长而略有降低。端位置取代也能对苝酰亚胺类衍生物的光物理性质实现微调。
虽然化合物PMD(1~4)具有较弱的导电性,但其仍具有一定的电致化学发光能力,在光导体、传感器等领域具有一定的应用价值。
