5-Aza-CdR干扰人子宫内膜样癌JEC细胞后ER拮抗剂对细胞增殖、形态及p57kip2蛋白表达的影响
2021-04-25张春英周正平
钟 栎,周 俊,张春英,周正平
(1.遵义医科大学附属医院 病理科,贵州 遵义 563099;2.遵义医科大学 电镜室,贵州 遵义 563099;3.遵义医科大学 病理学教研室,贵州 遵义 563099)
子宫内膜癌是发达国家最常见的妇科肿瘤,其发病率逐年上升[1]。据美国癌症协会估计,2019年在美国被诊断为子宫内膜癌的女性患者有61 880例,其中12 160例死于此病[2]。我国子宫内膜癌发病率约为60/10万,死亡率约为20/10万[3]。根据子宫内膜癌的致病机理和生物学行为特征,将其分为Ⅰ型和Ⅱ型。Ⅰ型的主要组织学类型为子宫内膜样癌(Endometrioid carcinoma,EC),其发病率约为80%~90%[4]。EC的发生发展主要与雌激素长期刺激有关[5]。雌激素与靶细胞雌激素受体(Estrogen receptor,ER)结合后,可通过核受体与膜受体两条途径,调节靶基因转录,促进细胞增殖[6]。ER拮抗剂分ER调节剂(Selective estrogen receptor modulator,SERM)和ER降解剂(Selective estrogen receptor degrading,SERD),SERM常使用他莫昔芬(Tamoxifen,TAM),TAM有Z型和E型两种构型,Z型与ER结合后表现抗雌激素作用,抑制细胞增殖,而E型则表现为弱雌激素样作用。SERD主要包括氟维司群(ICI-182780,ICI),ICI直接与ER结合并阻止ER的激活,发挥拮抗雌激素作用[7]。本课题组前期研究发现,TAM对JEC细胞有弱促增殖作用,ICI则有明显抗雌激素作用[8]。
EC的发生还与细胞周期的异常调控有关。p57kip2是细胞周期蛋白依赖性激酶(Cyclin-dependent-kinase,CDK)抑制剂(CKI)CIP/KIP家族的一员,可抑制细胞周期蛋白(Cyclin)D/E-CDK复合物活性,阻碍细胞从G1期向S期进展,抑制细胞增殖[9]。p57kip2在大多数肿瘤组织中,未见基因突变事件,异常表达可能与表观遗传改变中的DNA甲基化有关[10]。生理情况下,抑癌基因启动子常处于非甲基化状态,但由于其启动子区内富集的胞嘧啶-磷酸-鸟嘌呤(CpG)序列,故抑癌基因启动子较易发生甲基化[11]。癌基因的低甲基化、抑癌基因的高甲基化促进了肿瘤的发生发展[12]。5-氮杂-2'-脱氧胞苷(5-Aza-2'-deoxycytidine,5-Aza-CdR)是一种DNA甲基化转移酶(DNA Methyltransferase,DNMT)抑制剂,在低浓度下具有去甲基化作用,恢复抑癌基因的活性[13]。本实验拟使用亚硫酸氢盐测序(Bisulfite sequencing PCR,BSP)法检测5-Aza-CdR干扰前后JEC细胞p57kip2启动子区的甲基化率;分别用甲基偶氮唑盐比色(MTT)法、光镜和电镜、蛋白印迹(Western blot)法检测5-Aza-CdR干扰后TAM、ICI对JEC细胞的增殖、形态改变和p57kip2蛋白的表达情况的影响,进一步探讨p57kip2基因的DNA甲基化、ER拮抗剂与子宫内膜癌细胞增殖和分化的关系,为EC的内分泌辅助治疗提供一定的实验依据。
1 材料与方法
1.1 试剂与仪器 人EC中分化JEC细胞株[14]由遵义医科大学微生物教研室提供。雌二醇(β-estradiol,E2)、TAM、ICI购自Sigma公司,鼠抗人P57kip2单克隆抗体购自英国Abcom公司,鼠抗人β-actin单克隆抗体购自碧云天生物技术(上海)研究所。酶标仪(美国Thermoscientific公司),倒置显微镜CKX41(日本 OLYMPUS公司),透射电子显微镜H-7650(日本日立公司),Western blot相关仪器(BIO-RAD公司)。
1.2 细胞培养 配制完全培养液(10%胎牛血清、100 U/mL青霉素、100 U/mL链霉素、90%RPMI 1640培养液)体外培养JEC细胞,培养环境为37 ℃、5%CO2。
1.3 方法
1.3.1 BSP法检测JEC细胞p57kip2启动子区的甲基化率 分别用含0 mol/L、1.25×10-5mol/L的5-Aza-CdR的培养基培养JEC细胞24 h后,经胰酶消化后离心成细胞团块,送生工生物工程(上海)股份有限公司提取DNA后通过BSP法分别检测5-Aza-CdR(0 mol/L)组、5-Aza-CdR(1.25×10-5mol/L)组细胞p57kip2启动子区32个CPG位点的甲基化状态。引物序列为:Forward,5’-GTTAGTAGGTGTGTGAGGGTTTTAG-3’,Reverse:5’-CTCCTTTATCTACAAACRAAAACCTC-3’,扩增长度298 bp。PCR反应条件:95 ℃预变性3~5 min,94 ℃变性30 s,55~60℃变性30 s,72 ℃退火30~50 s,循环35次,最后72 ℃延伸5~8 min后测序。
1.3.2 检测5-Aza-CdR干扰后ER拮抗剂对JEC细胞的影响
1.3.2.1 实验分组 将JEC细胞分为对照组、E2组、TAM组、ICI组、E2+TAM组、E2+ICI组,各组药物的终浓度均为1.0×10-6mol/L。使用等量且浓度为1.25×10-5mol/L的5-Aza-CdR预先处理各组细胞24 h后,体外继续培养各组24、48、72 h;72 h时段的各组细胞在培养48 h时追加一次等量且浓度为1.25×10-5mol/L的5-Aza-CdR。
1.3.2.2 MTT法检测JEC细胞的增殖活性 取对数生长期的JEC细胞,制成单细胞悬液(1.0×105个/mL)后接种于96孔板中,每孔100 μL,每组设 5个复孔。经上述药物处理,分别培养各组24、48、72 h后,弃培养液,PBS溶液冲洗后各孔加入5 mg/mL的MTT溶液20 μL(避光操作),放入培养箱中充分反应4h,弃MTT溶液后每孔再加入100 μL DMSO,避光振荡10 min,在酶联免疫检测仪波长490 nm处检测各孔的OD值。OD值越大,活细胞数量越多,细胞的增殖能力越强。实验重复3次。
1.3.2.3 倒置显微镜观察JEC细胞的生长情况 6孔板培养JEC细胞,每组设3个复孔。经上述药物处理,培养各组72 h后,通过倒置显微镜观察细胞的密度和形态的改变。实验重复3次。
1.3.2.4 透射电镜观察JEC细胞的超微结构 经上述药物处理,培养各组72 h后,经胰酶消化后高速离心成细胞团块,加入固定液(2.5%戊二醛)固定,经脱水、浸透、包埋聚合,用超薄切片机行超薄切片、电子染色后,在透射电镜下进行观察。
1.3.2.5 Western blot法检测JEC细胞p57kip2蛋白的表达情况 经上述药物处理,培养各组72 h后,加入RIPA裂解液200 μL/孔,冰上裂解0.5 h,超速离心后提取蛋白,行BCA法定蛋白浓度。配制分离胶及浓缩胶(浓度分别为12%、5%),行SDS-PAGE分离蛋白;将分离后的目的蛋白所在区域的凝胶切下,转移至PVDF膜上后,摇床上用封闭液(5%脱脂奶粉)封闭2 h,加入一抗[鼠抗人P57kip2和β-actin单克隆抗体(体积稀释比例分别为1∶200和1∶1 000)]孵育过夜,经TBST洗膜3次后加入二抗[兔抗鼠IgG(体积稀释比例为1∶10 000)],室温下充分反应2 h,再用TBST洗膜3次,最后用Odyssey系统将PVDF膜扫描成像后检测蛋白条带的D值。以目的条带与内参照β-actin条带灰度值之比表示目的蛋白的相对表达水平。实验重复3次。

2 结果
2.1 BSP法检测JEC细胞p57kip2启动子区甲基化率的结果 培养24 h后,JEC细胞p57kip2启动子区甲基化率在5-Aza-CdR(0mol/L)组为88.1%,在5-Aza-CdR(1.25×10-5mol/L)组为76.2%(见图1)。
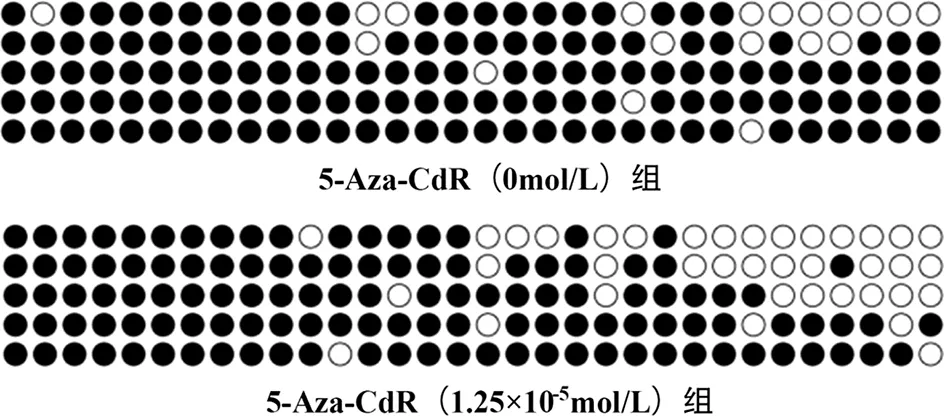
图1 5-Aza-CdR(0 mol/L)组和5-Aza-CdR(1.25×10-5mol/L)组JEC细胞p57kip2启动子区的甲基化率
2.2 5-Aza-CdR干扰后ER拮抗剂对JEC细胞的影响
2.2.1 MTT检测结果 与对照组相比,E2组和TAM组的细胞增殖活性在24、48、72 h均升高,其中E2组在24 h升高更显著(P<0.05);ICI组、E2+TAM组、E2+ICI组在各时间段的细胞增殖活性均下降,且均在24 h下降更明显(P<0.05)。与E2组相比,ICI组、E2+TAM组、E2+ICI组的细胞增殖活性在各时间段均下降,其中以E2+TAM组在24、48 h下降最明显(P<0.05),E2+ICI组在72 h下降最显著(P<0.05)。在同一药物分组下:与24 h比较,对照组、TAM组、ICI组、E2+TAM组的细胞增殖活性在48 h升高(P<0.05),E2组、TAM组、E2+TAM组、E2+ICI组的细胞增殖活性在72 h升高不明显(P<0.05,见图2)。
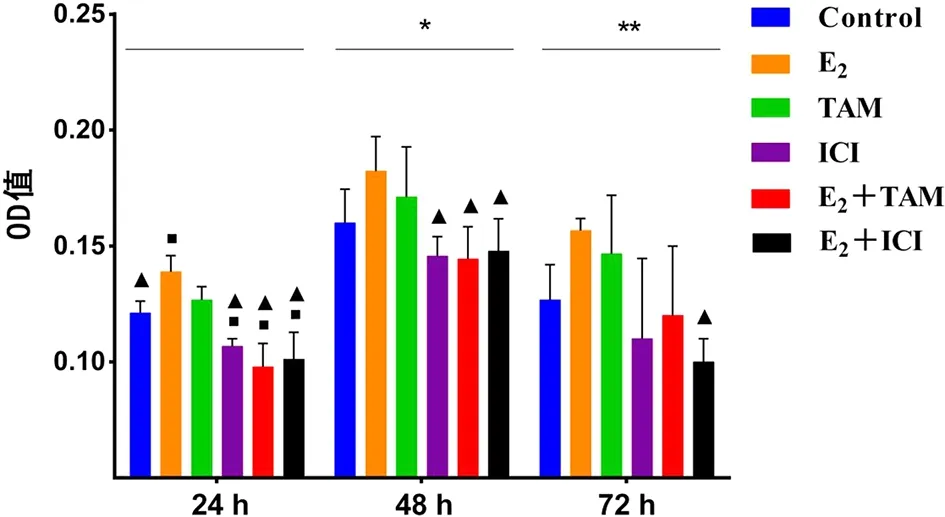
■:P<0.05 vs 对照组;▲:P<0.05 vs E2组;*:P<0.05 vs 24 h 对照组、TAM组、ICI组、E2+TAM组;**:P<0.05 vs 24 h E2组、TAM组、E2+TAM组、E2+ICI组。
2.2.2 倒置显微镜观察JEC细胞的生长情况 培养72 h,对照组细胞均匀布满视野的70%~80%,细胞形态呈梭形或不规则形,可见围腺腔样结构。与对照组相比,E2组细胞密度明显升高,TAM组稍增加,ICI组、E2+ICI组、E2+TAM组细胞密度均降低,其中以E2+TAM组下降最明显(见图3)。
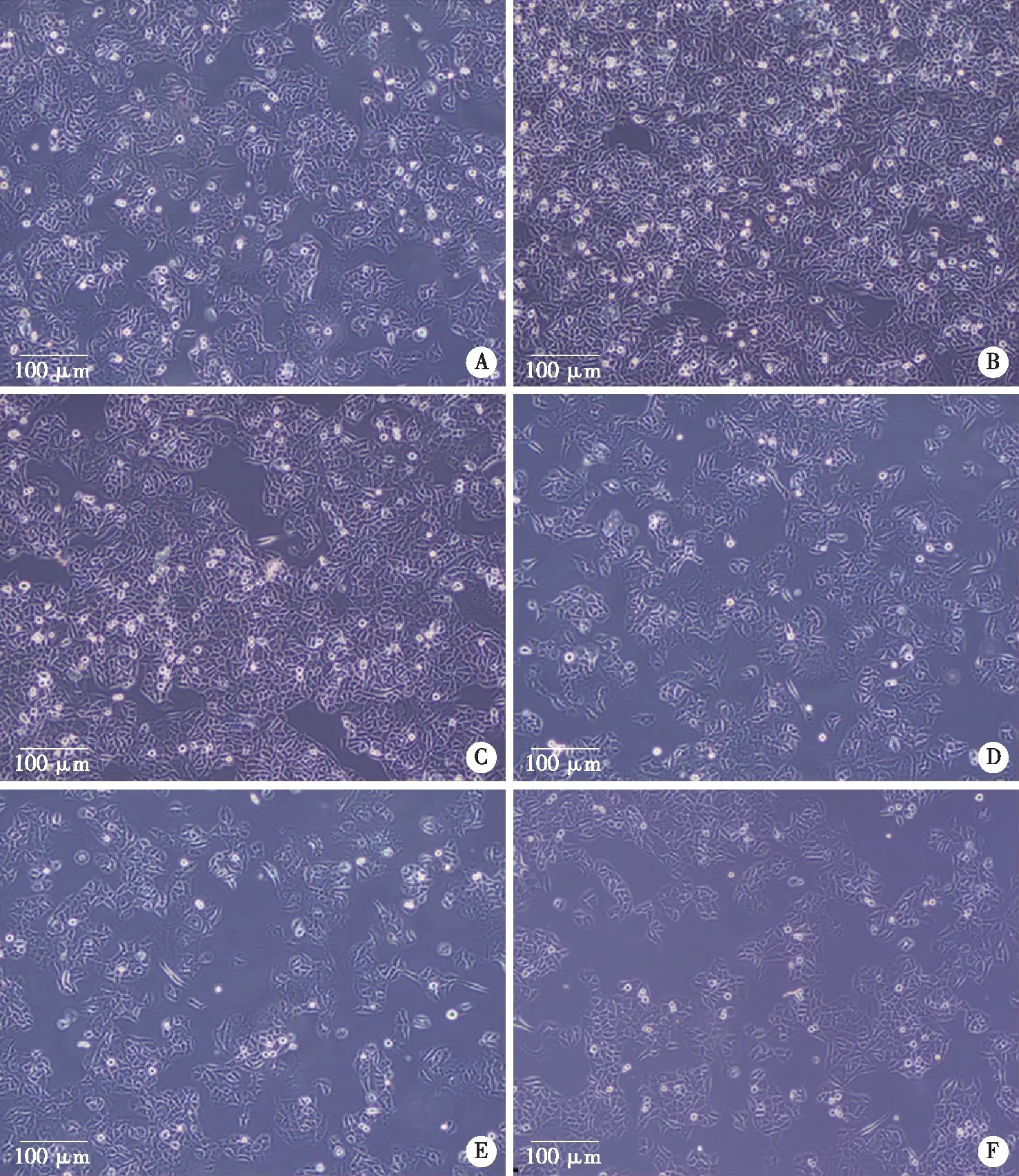
A:对照组;B:E2组;C:TAM组;D:ICI组;E:E2+TAM组;F:E2+ICI组。
2.2.3 透射电镜观察JEC细胞的超微结构 培养72 h,对照组细胞呈圆形或类圆形,细胞游离缘可见少量微绒毛;细胞内可见一定数量的线粒体,部分细胞内可见扩张的内质网和自噬溶酶体,偶见分泌泡;细胞核大而不规则,核仁明显;与对照组比较,E2+TAM组细胞游离缘微绒毛稍增多,其余各实验组细胞游离缘微绒毛均有所减少;各实验组细胞内均可见较为丰富的线粒体及稍扩张的内质网,可见少量自噬溶酶体散在分布于胞质内;TAM组、ICI组、E2+TAM组、E2+ICI组细胞内分泌泡增多,其中以E2+TAM组、E2+ICI组分泌泡数量最多;TAM组、E2+ICI组部分细胞可见凋亡小体(见图4)。
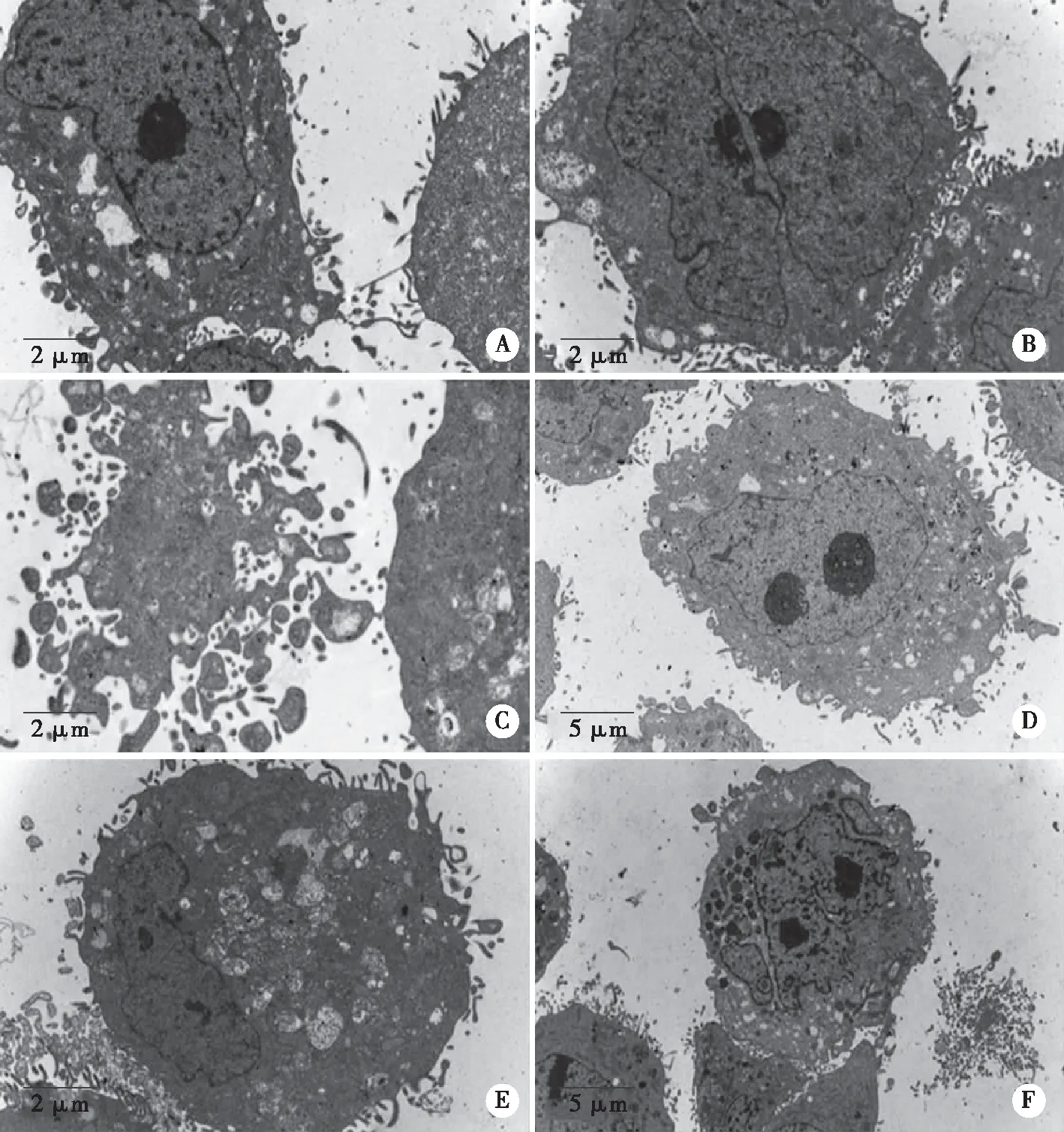
A:对照组(Bar=2.0 μm);B:E2组(Bar=2.0 μm);C:TAM组(Bar=2.0 μm);D:ICI组(Bar=5.0 μm);E:E2+TAM组(Bar=2.0 μm);F:E2+ICI组(Bar=5.0 μm)。
2.2.4 Western blot结果 培养72 h,与对照组相比,E2组的p57kip2蛋白表达减少(P<0.05);ICI组、E2+TAM组、E2+ICI组中p57kip2蛋白的表达量均明显增高(P<0.05),其中E2+TAM组增高最明显(P<0.05);TAM组的p57kip2蛋白的表达量略高于对照组(P>0.05,见图5~6)。
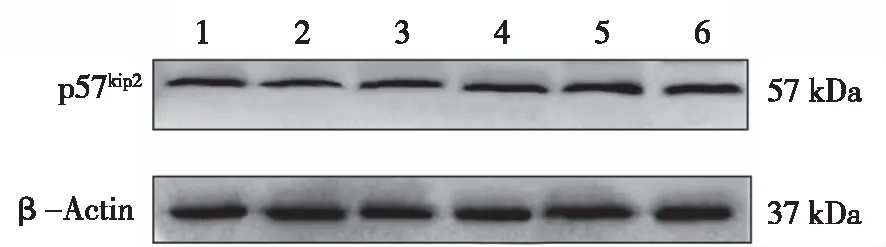
1:对照组;2:E2组;3:TAM组;4:ICI组;5:E2+TAM组;6:E2+ICI组。

★:P<0.05 vs 对照组;◆:P<0.05 vs E2组。
3 讨论
子宫内膜癌是最常见的女性生殖系统恶性肿瘤之一,在我国的发病率仅次于宫颈癌,严重威胁女性健康[3]。子宫内膜癌的主要病理类型是EC,其发生发展除无孕激素拮抗的雌激素长期刺激外[5,15]还与基因突变[16]、细胞周期调控异常和表观遗传改变等因素有关[17]。
研究表明,对已接受乳腺切除术的乳腺导管原位癌患者使用TAM治疗后会出现子宫内膜增生、子宫内膜癌等不良反应,这可能与TAM的组织选择性有关,在乳腺组织可拮抗ER发挥拮抗雌激素作用,而在子宫内膜组织中激活ER发挥雌激素样作用[18]。ICI对ER具有很高的亲和力,与ER结合后能完全阻断ER的转录活性并降解ER,从而发挥抗雌激素作用,而不诱导子宫内膜增殖(无类雌激素样作用)[7]。
p57kip2是定位于11p15.5的抑癌基因,是CIP/KIP家族成员中最晚被发现的。p57kip2蛋白由316个氨基酸组成,其氨基末端与Cyclin-CDK复合物结合后,能抑制CDK活性,负向调控细胞周期[9]。研究发现,p57kip2蛋白在淋巴瘤、乳腺癌、前列腺癌、肺癌[19-22]等组织中的表达明显下调或缺乏。
p57kip2是父系等位基因沉默,母系等位基因表达的印记基因,其表达的下调或缺乏可能与表观遗传修饰相关[10]。表观遗传修饰常为DNA甲基化、组蛋白乙酰化[23]。DNA甲基化指甲基(CH3)基团在DNMT的催化下共价结合到DNA序列上的胞嘧啶残基上,生成5-甲基胞嘧啶的过程,整个过程不改变DNA序列,而只对部分碱基对进行甲基化修饰[11]。DNA的过度甲基化是引起抑癌基因失活的重要方式之一。目前许多研究证明肿瘤抑癌基因的失活与该基因的启动子区的CpG岛高甲基化状态有关[24]。在肺鳞癌和早期声带癌的研究中发现p57kip2基因启动子区的高甲基化与p57kip2蛋白不表达或低表达有关[25-26]。
5-Aza-CdR是一种胞嘧啶类似物,能代替胞嘧啶捕获DNMT并掺入DNA中,随细胞的分裂,导致DNMT降解,从而降低肿瘤细胞中抑癌基因启动子区的高甲基化水平,解除高甲基化对抑癌基因生物学功能的抑制,使抑癌基因重新表达或使其表达上调,诱导肿瘤细胞凋亡或向正常细胞分化[13]。已有研究显示,5-Aza-CdR能逆转RASSF1A基因高甲基化状态,使因高甲基化而完全失活的RASSF1A基因重新表达,抑制子宫内膜癌HEC-1-B细胞的增殖[27]。本实验研究结果表明,1.25×10-5mol/L的5-Aza-CdR干扰JEC细胞后,可以使其p57kip2启动子区高甲基化水平明显下调。由于DNA甲基化不是遗传学上的DNA序列改变,而是一种是可逆的表观遗传学改变,因此5-Aza-CdR可有效逆转p57kip2启动子区的高甲基化状态,可能使因p57kip2启动子区高甲基化而低表达的p57kip2蛋白上调到相对较高水平,恢复p57kip2作为肿瘤抑制因子的生物学效应,负调控细胞周期,抑制肿瘤细胞增殖。
目前研究显示,5-Aza-CdR能增强化疗药物在表皮样癌、膀胱癌中的敏感性[28-29]。本课题组前期研究结果显示:TAM可微弱促进JEC细胞增殖,ICI能抑制JEC细胞增殖,E2+TAM组、E2+ICI组细胞增殖能力高于对照组[8]。而本实验结果显示5-Aza-CdR干扰JEC细胞后,ICI组、E2+TAM组、E2+ICI组细胞的增殖能力明显被抑制。表明5-Aza-CdR干扰后,能增强JEC细胞对TAM的敏感性,使TAM发挥对JEC细胞增殖的抑制作用,并加强ICI对JEC细胞增殖的抑制,这可能与5-Aza-CdR能逆转JEC细胞p57kip2启动子的高甲基化状态,从而恢复p57kip2负调控细胞周期的作用有关;同时5-Aza-CdR可能促进相关肿瘤特异性抗原及肿瘤特异性免疫刺激分子的分泌,降低JEC细胞的耐药性,从而提高JEC细胞对ER拮抗剂的敏感性。此外,E2+TAM能明显抑制JEC细胞增殖,可能还与5-Aza-CdR使因启动子高甲基化而沉默的ERɑ基因重新表达有关。Tang等[30]研究发现,5-Aza-CdR可使因启动子高甲基化而沉默的ERɑ基因重新表达,诱导ERɑ阴性乳腺癌表达ERɑ蛋白,恢复ERɑ阴性乳腺癌细胞对TAM的敏感性。而本课题组前期研究表明,JEC细胞不表达ERɑ蛋白[8],推测JEC细胞中ERɑ蛋白的不表达可能与ERɑ基因启动子高甲基化有关,5-Aza-CdR可能通过逆转ERɑ基因启动子高甲基化状态,使沉默的ERɑ基因重新表达,增强TAM在EC中的敏感性。
在细胞形态结构上,本课题组前期研究结果显示:JEC细胞内分泌泡不明显,ICI组可见自噬现象[8]。而本实验研究结果显示:5-Aza-CdR干扰JEC细胞后,各组细胞内均可见扩张的内质网、自噬溶酶体及一定数量的分泌泡,其中以E2+TAM组、E2+ICI组分泌泡数量最多,提示细胞分泌功能增强;对照组、TAM组、E2+ICI组部分细胞可见凋亡现象。表明5-Aza-CdR不仅可使因启动子高甲基化而呈表观抑制的p57kip2恢复其抑癌因子的功能,抑制肿瘤细胞生长;其本身可能还具有促进肿瘤细胞向正常细胞分化或诱导肿瘤细胞凋亡以及使肿瘤细胞的分泌功能增强的作用。同时5-Aza-CdR干扰JEC细胞后,细胞内可见扩张的内质网,提示5-Aza-CdR可能对细胞有一定损伤作用。有文献报道,高浓度的5-Aza-CdR能直接抑制DNA的合成而促进细胞死亡,产生细胞毒作用,而低浓度的5-Aza-CdR可使DNMT失活,除去抑癌基因启动子区的高度甲基化状态,抑制肿瘤细胞生长,并且这种去甲基化作用具有可遗传性[31]。有研究证明,10μg/mL的5-Aza-CdR对纤维肉瘤细胞中的DNA、RNA及蛋白质合成没有明显的抑制作用[32]。而本实验中用1.25×10-5mol/L的5-Aza-CdR干扰JEC细胞,主要为对其抑癌基因p57kip2启动子的去甲基化作用,而非抑制DNA的合成的药物细胞毒作用。
在p57kip2蛋白表达水平上,本课题组前期研究结果显示:与对照组比较,TAM组和E2+ICI组中p57kip2蛋白的表达水平微弱上调,ICI组p57kip2蛋白的表达水平明显上升,E2+TAM组p57kip2蛋白的表达水平无明显改变[8]。本实验的结果显示:5-Aza-CdR干扰后,TAM、ICI、E2+ICI、E2+TAM均能上调p57kip2蛋白的表达水平,且TAM、ICI分别与E2的联合使用比单独使用TAM、ICI能更为显著上调p57kip2蛋白的表达。表明DNA高甲基化所致抑癌基因p57kip2表达下调是可逆转的,通过5-Aza-CdR逆转p57kip2启动子区的高甲基化状态,可能恢复p57kip2基因的活性,上调p57kip2蛋白的表达水平,促进肿瘤细胞凋亡、抑制细胞增殖;5-Aza-CdR还可能协同TAM、ICI上调p57kip2蛋白的表达水平,拮抗E2对p57kip2蛋白水平的下调,达到削弱CDK的激酶活性,从而发挥负向调控细胞周期、抑制JEC细胞进一步生长的作用。
