基于转录组测序的越南安息香根、茎和叶基因表达分析
2021-04-22王小敏许婳婳詹若挺马新业
王小敏,许婳婳,詹若挺,马新业*
• 药材与资源 •
基于转录组测序的越南安息香根、茎和叶基因表达分析
王小敏1,许婳婳2,詹若挺1,马新业1*
1. 广州中医药大学 中药资源科学与工程研究中心,岭南中药资源教育部重点实验室,国家中成药工程技术研究中心 南药研发实验室,广东 广州 510006 2. 广州中医药大学中药学院,广东 广州 510006
对越南安息香进行转录组测序,获得其根、茎和叶的转录组信息特征。以越南安息香的根、茎和叶作为研究对象,使用Illumina HiSeqTM2000进行越南安息香根、茎和叶的转录组测序分析。转录组测序根、茎和叶共获得53 835 045条高质量序列(clean reads),Trinity de novo组装获得69 151条Unigenes,平均长度778.51 nt。BLAST分析表明分别有41 412(59.89%)、31 189(45.10%)、25 539(36.93%)、16 749(24.22%)个Unigenes在Nr、Swiss-port、KOG、KEGG数据库中得到注释,可归入GO分类的细胞组分、生物过程和分子功能3大类46分支,涉及129条KEGG标准代谢通路,其中有31个次生代谢标准通路。蛋白编码框序列3 461个,涉及高等植物转录因子54个家族。使用MISA软件挖掘10 974个简单重复序列(SSRs),二碱基重复SSRs数量最为丰富,有6282个(57.24%),五碱基重复SSRs最少,占2.45%。利用高通量技术和生物信息分析获得了越南安息香根、茎和叶的转录组信息特征,为后期越南安息香基因功能鉴定、次生代谢途径解析及调控机制的研究奠定基础。
越南安息香;转录组;功能基因;代谢通路;简单重复序列
越南安息香(Pierre) Crail ex Har-tw.为安息香科(Styracaceae)安息香属落叶乔木,又名越南安息香、白背安息香和滇桂野茉莉,俗称“东京野茉莉”,广泛分布于我国岭南地区,老挝、缅甸等国也有分布;其茎皮受到伤害后的分泌物干燥后作为安息香[1],始载于《新修本草》,味辛、苦,平,无毒;主心腹恶气鬼疰。安息香的现代药理学认为安息香具有抗细菌[2]、抗真菌[3]、抗补体[4]、抗氧化[5]、抗白血病[6]、抗肿瘤[7]等药理活性。现代中药学揭示安息香含有香脂酸类成分,包括苯甲酸、松柏树乙醚、香草醛、苯甲酸苄脂、2-丙炔酮[8],此外,安息香含有三萜类成分,包含苏门答刺树脂酸、泰安息香树脂酸、齐墩果酸[6],可为药物研发提供丰富的化合物前体。因此,安息香的资源开发和基础研究具有重要的价值和前景。
转录组测序对于没有基因组的物种来说,可以有效地表征和鉴定植物中次生代谢物质的生物合成相关途径,揭示植物体的生长发育、生理适应性以及探索其中的基因序列和表达水平[9-11]。近年来,Illumina测序技术已广泛应用于研究植物基因组,并且也应用在番茄[12]、人参[13]、扁豆[14]等物种中,为其种植资源和基因遗传打下基础。本研究选择越南安息香幼苗的根、茎和叶作为基础材料,使用Illumina HiSeq-2000采集RNA-seq读数,以期挖掘安息香根、茎和叶的整体基因表达特征,为该植物基因功能研究、次生代谢途径解析和调控探究提供数据基础。
1 材料与方法
1.1 RNA提取与测序
植物材料于2015年10月采自广州中医药大学,经广州中医药大学马新业副研究员鉴定为越南安息香(Pierre) Crail ex Har-tw.。取单株植物根、茎、叶液氮冷冻后迅速存于−80 ℃冰箱至使用。
越南安息香幼苗根、茎和叶的总RNA分离后,用含有Oligo(dT)的磁珠富集mRNA,加入fragmentation buffer使mRNA成为短片段,合成cDNA第1条链使用的是六碱基随机引物(random hexamers),使用添加EB缓冲液的QiaQuick PCR试剂盒纯化并洗脱,经末端修复、加测序接头、加poly(A),大小片段的回收使用琼脂糖凝胶电泳,最后通过PCR扩增完成cDNA文库制备,由广州基迪奥生物科技有限公司(广州)使用Illumina HiSeqTM2000进行测序。
1.2 序列的从头组装和功能注释
采用FastQC软件(http://www. bioinformatics. babraham.ac.uk/projects/fastqc/)评估原始数据的质量,NGS QC Toolkit(v2.3.3)软件舍弃低质量读取(http://59.163.192.90:8080/ngsqctoolkit/)。使用Trinitysoftware[11]对RNA-Seq从头组装获得Unigene,其总体表达量使用RPKM法[15]计算。
为了注释转录组,利用BLAST将Unigenes比对到蛋白数据库Nr、Swiss-prot、蛋白相邻类的聚簇(KOG)和京都基因与基因百科全书(kyoto encyclopedia of genes and genomes,KEGG)(值<1×10−5),获得与相应Unigenes具有最高相似性的蛋白,从而得到相应的注释,使用Blast2GO软件对Unigenes进行GO(gene ontology)分类, 用WEGO软件对获得的Unigenes进行GO分类,宏观上认识该物种基因功能的分布情况。
1.3 蛋白编码框(sequence coding for aminoacids in protein,CDS)及转录因子的鉴定
按照Nr、Swiss-Prot、KOG和KEGG优先顺序把Unigenes序列和以上数据库进行BLASTx比对(<1×10−5),并确定该Unigene编码区的核苷酸序列(序列方向5’至3’端)及氨基酸序列。使用ESTScan预测与以上数据库对比不上的Unigenes的编码区和序列方向。将预测到的Unigenes编码蛋白序列和植物转录因子数据库plantTFDB进行hmmscan比对,获得转录因子家族和相关成员。
1.4 简单重复序列(simple sequence repeats,SSRs)特征检测
MISA软件被用到越南安息香转录组Unigenes的检测,搜索SSRs同时进行统计分析。
2 结果与分析
2.1 转录组组装和质量评估
通过Illumina HiSeqTM2000测序,越南安息香的根、茎和叶分别获得65 475 824、66 555 768、56 770 948条raw reads,滤过后获得64 267 196(98.15%)、65 122 652(97.85%)、55 714 504(98.14%)条clean reads,分别包含滤过后核苷酸8 033 399 500、8 140 331 500和6 964 313 000个。Q20(碱基量≥20%)均大于98%和Q30(碱基量≥30%)均>96%,说明测序数据控制良好,clean reads质量合格。Trinity组装得到69 151条Unigenes,平均长度778.51 nt,最长达10 486 nt,最短为201 nt,N50等于1333。
2.2 Unigenes功能注释
用BLAST将所获得的Unigenes比对到Nr、Swiss-port、KOG、KEGG数据库,并统计所注释到的Unigenes数目和功能信息。结果显示,41 412(59.89%)条Unigenes在Nr数据库得到注释,在Swiss-prot、KOG和KEGG数据库分别注释了31 189(74.3%)、25 539(60.9%)、16 749(39.9%)条Unigenes。成功注释的Unigenes共41 951条,占66.67%,27 200(39.33%)条未得到注释。Unigenes长度分布见图1-A,17 387条Unigenes长度大于等于1000 nt,5674条Unigenes超过2000 nt。图1-B展示的是reads在Unigene上的覆盖度,reads在11~100的Unigenes数量最多,有27 240条;其次是reads在1~10的Unigenes,有5150条;reads在101~200、2001~4000、1001~2000、>10 000的数量分别是4669、4226、3935和3186条;其他reads在Unigenes上的覆盖度均相对较小。
以注释到Nr数据库的Unigenes为例,同源物种如图2,在匹配度最高的物种中,葡萄L.占的比率为最高,达4160(10.05%)条;其次为欧洲油菜L. 1889(4.56%),黄豆(Linn.) Merr. 1630(3.94%),毛果杨L. 1615(3.90%),蒺藜苜蓿L. 1614(3.90%),拟南芥L. 1577(3.81%),粳稻Japonica Group 1563(3.77%),可可树L. 1537(3.71%),番茄L. 1400(3.38%),苹果B. 1262(3.05%);匹配度在3.04%~1.46%的有13 304条,小于1.46%匹配度的有9 861条,占23.81%。根据Nr进一步获得GO分类信息,如图3,148 884条Unigenes被注释到细胞组分、生物过程和分子功能3个GO类别的46个小组。生物过程中代谢过程(metabolic process)、细胞过程(cell process)和多细胞生物过程(multicellular organismal process)丰度最高,分别为15 930、14 748和11 806 条;生物调节(biological regulation)和应激适应(response to stimulus)分别有5087和4630条。分子功能中催化活性(catalytic activity)和结合功能(binding)的基因最多,分别达15 160和14 029条,其他基因表达量都较少。细胞组分中细胞(cell)及细胞组分(cell part)较高,分别达10 915和10 914条;细胞器(organelle)、细胞膜(membrane)分别有8509和5938条;细胞膜组分(membrane part)、器官组分(organelle part)和大分子复合物(macromolecular complex)分别有4125、3713和2712条,其他基因均较小。
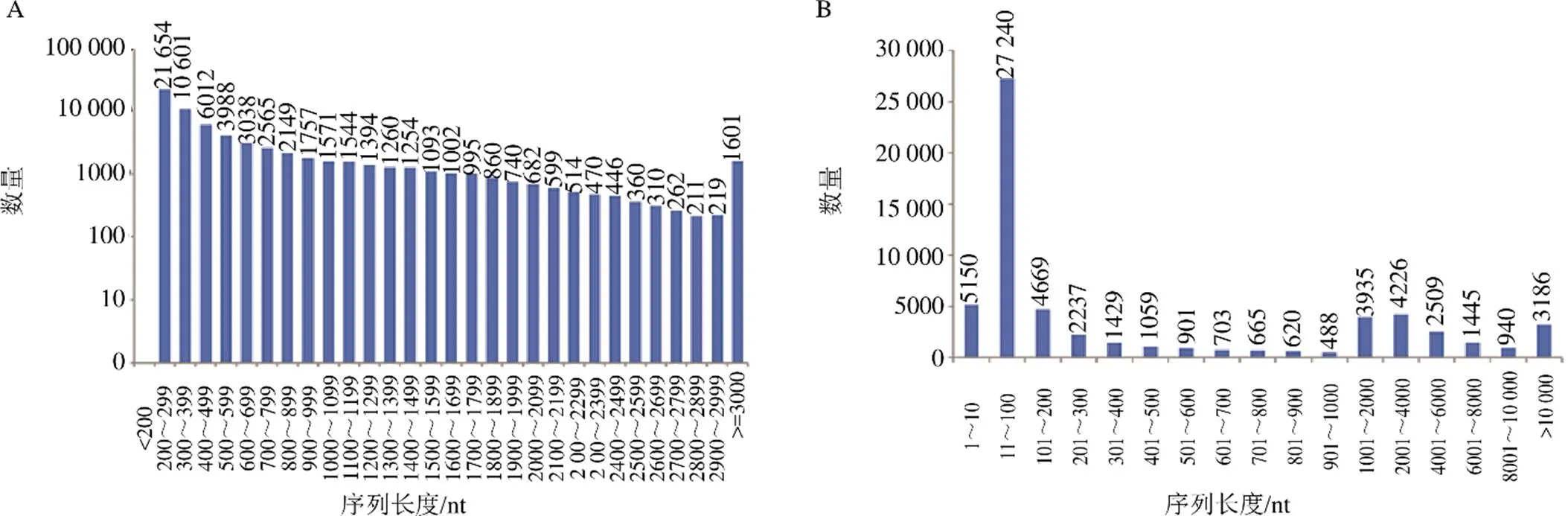
图1 Unigenes的长度分布(A) 和reads的覆盖范围(B)

图2 Unigenes在Nr数据库中的物种匹配
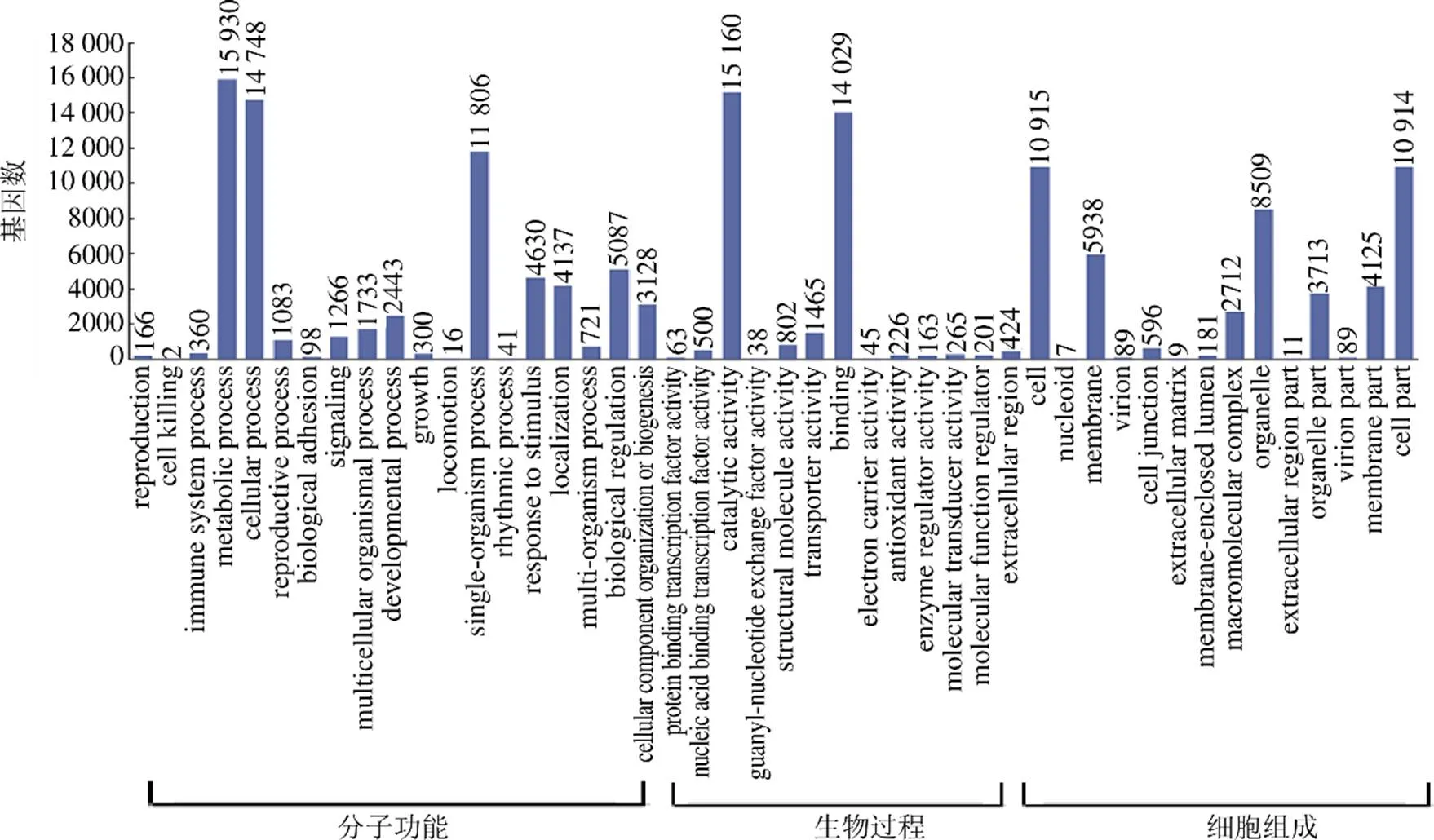
图3 转录组Unigenes的GO分布
为了进一步分析越南安息香的转录组Unigenes的功能,使用KOG功能分类分析(图4),一共获得25种KOG分类,种类较全面,其中,一般功能预测的基因(general function prediction only)达8 025条,为最多;信号转导机制类(signal transduction mechanisms)、翻译后修饰、蛋白反转和伴侣(posttranslational modification, protein turnover, chaperones)次之,分别有5082和4862条;其他分类基因丰度参差不齐。
越南安息香根、茎和叶的转录组Unigenes参与KEGG代谢通路被分为5类:代谢(metabolism)8878条、遗传信息处理(genetic information processing)3 936条、细胞过程(cellular processes)753条、有机系统(organismal systems)573条和环境信息处理(environmental information processing)548条。其中,7533条Unigenes被归类于94个标准代谢通路,根据注释量选择前9个代谢通路展示见表1,这些通路含有的Unigenes数量均不低于200条。

图4 转录组Unigenes的KOG分布
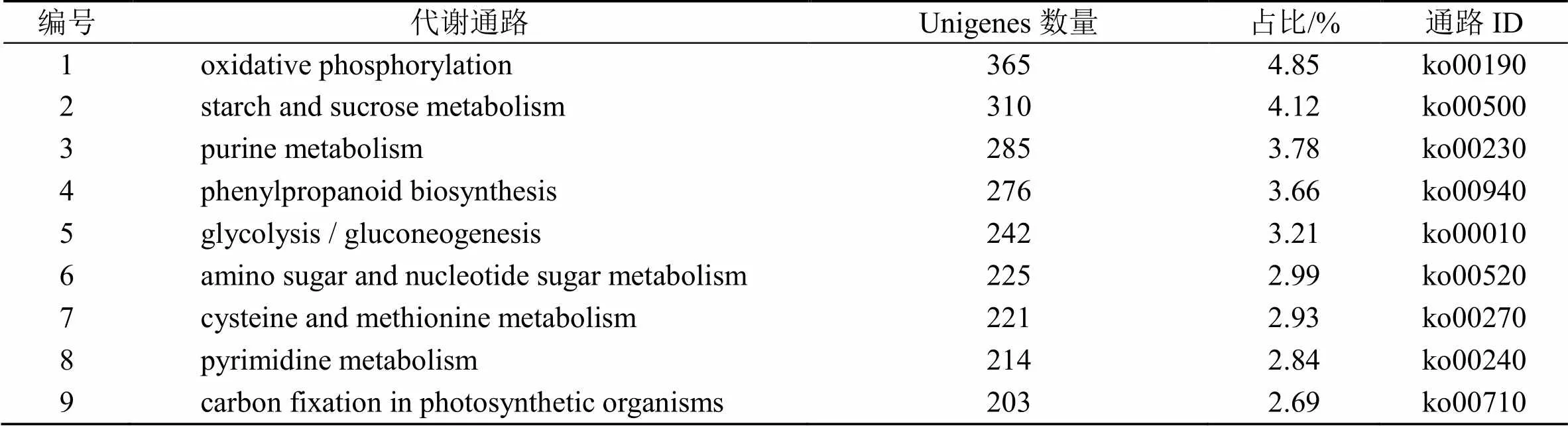
表1 转录组Unigenes的KEGG通路统计分析
进一步分析KEGG发现,940条Unigenes参与苯丙烷类、类黄酮类、萜类等20个次生代谢标准生物合成通路(表2)。其中,苯丙烷类生物合成(ko00940)注释到的基因数最多,达276条;其次是类黄酮的生物合成(ko00941),基因数为100条;萜类化合物骨架生物合成(ko00900)有关基因93条;80条基因与芪类化合物、二芳基庚烷和姜辣素(ko00945)有关;分别有64、46、45和41条基因与类胡萝卜素生物合成(ko00906)、异喹啉生物碱生物合成(ko00950)、二萜类生物合成(ko00904)和莨菪烷类、哌啶和哌啶生物碱生物合成(ko00960)有关;37条参与玉米素的生物合成(ko00908);油菜素类固醇的生物合成(ko00905)有35条;34条基因涉及倍半萜和三萜类的生物合成(ko00909);花青素、单环内酰胺类、柠檬烯和蒎烯的降解、单萜类生物合成、黄酮和黄酮醇类合成、异黄酮类合成、芥子油苷的生物合成、咖啡因的合成和甜菜红色素的生物合成相关的基因均较少,在30条以下。这些数据为越南安息香次生代谢机制研究提供数据基础。
2.3 CDS和转录因子分析
对越南安息香所有Unigenes进行CDS分析,通过BLAST获得41 351个CDS序列,利用ESTscan数据库得到3461条CDS。转录因子通过预测,被分到54个转录因子家族,其中C2H2、ERF、bHLH、MYB、GRAS、NAC、MYB-relate和WRKY占主体,说明众多转录调控过程(图5)参与了越南安息香根、茎和叶的生理代谢。

表2 转录组Unigenes次生代谢的KEGG通路注释统计
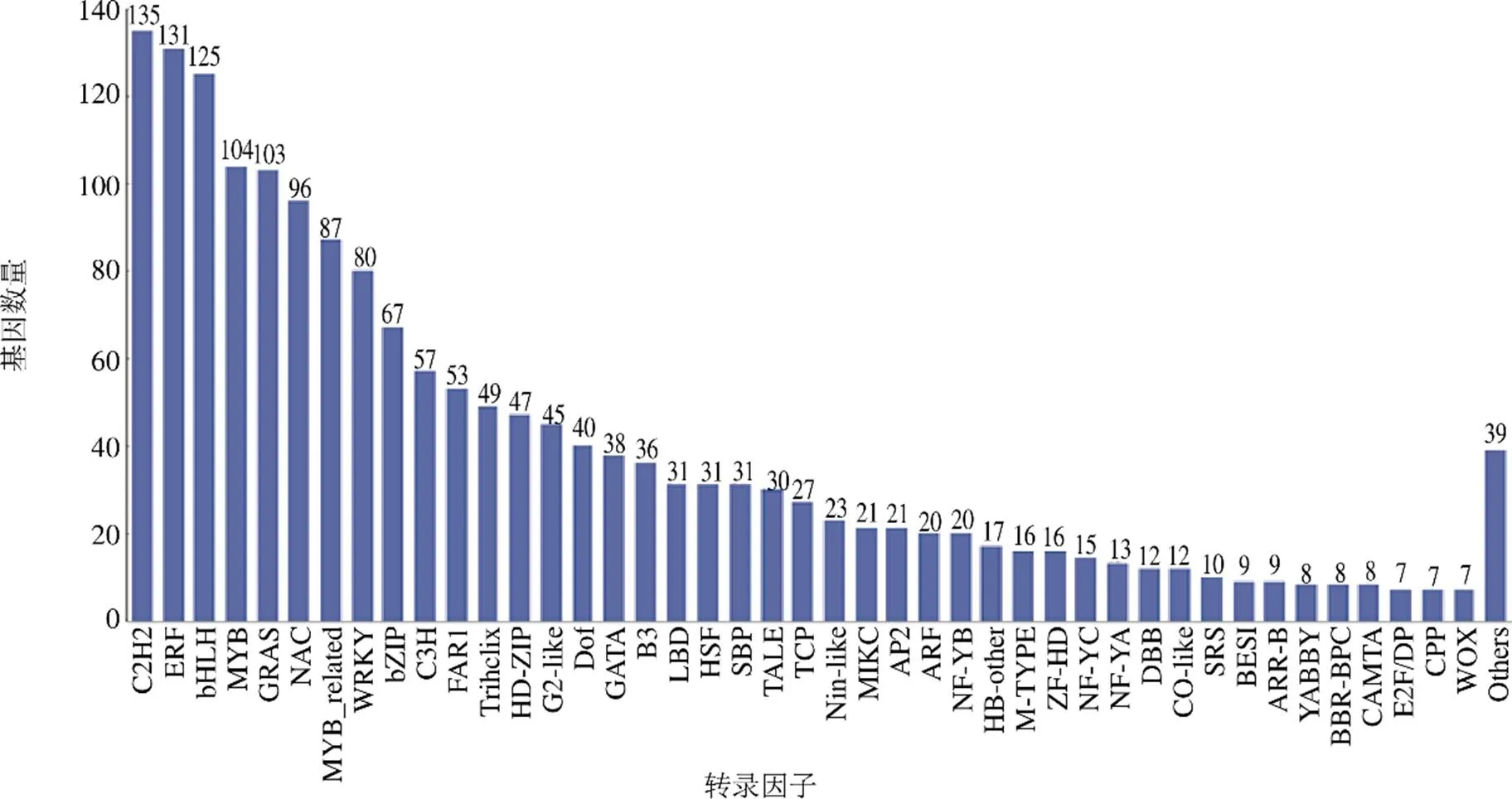
图5 转录因子分析
2.4 SSRs特征分析
MISA软件用于Unigenes的SSRs检索,8712个Unigenes中有10 974个SSRs(表3)。二碱基重复SSRs丰度最高,为6282(57.24%)个,其中,AG/CT类型所占比例最高;其次是三碱基重复,3072(27.99%)个;四、五和六碱基分别有777(7.08%)、269(2.45%)和574(5.23%)个。
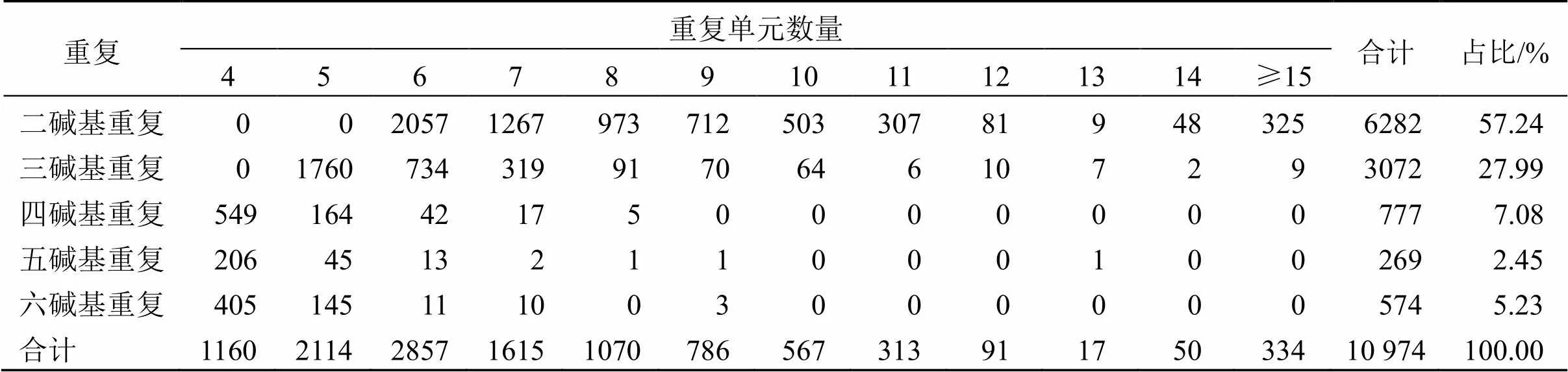
表3 转录组Unigenes SSRs分布
3 讨论
高通量测序技术在药用植物的研究上已经展开了较为广泛的应用,并取得了重大的进展[16]。该研究首次对越南安息香采用Illumina HiSeqTM2000技术获得其转录组数据并进行相关分析,测序质量与质控结果均良好,53 835 045条clean reads组装得到69 151条Unigenes,其长度与reads覆盖度均合理。Unigenes数据巨大,基本涵盖所有转录组信息,初步揭示越南安息香根、茎和叶的基因表达特征。
高通量测序获得的转录组数据的相关分析与生物信息学分析密切关联。本研究通过生物信息学分析对所获得的转录组数据进行reads滤过、组装,以及基因注释和功能的分类等,涉及的软件有BLAST、Trinity、ESTscan等。
基于BLAST分析,69 151条Unigenes通过4大数据库(Nr、KEGG、KOG和Swiss-port)比对有41 951(60.67%)条被成功注释,该注释占比与已经报道的甘草[17]、黄精[18]、地黄[19]等物种类似,反映了越南安息香中存在大量未知序列特征及功能的Unigenes,有待进一步探究。
越南安息香转录组的GO分类揭示其特性与细胞组分、细胞过程和分子功能相关;通过KOG数据库从基因水平寻找直系同源物种,预测ORF的生物学功能,很大程度上提高了注释的准确度[20],该研究获得较为全面的KOG类群(25个)。进一步通过KEGG数据库对越南安息香转录组分析,发现了94个标准代谢通路,这些通路可能与越南安息香的水分和矿物质吸收、光合和呼吸作用等生理过程相关。此外,还发现20条次生代谢标准通路,涉及了苯丙烷类、黄酮类及萜类等生物合成。其中,参与苯丙烷类生物合成、萜类生物合成的Unigenes分别有276个和91个。越南安息香的次生代谢产物以苯丙烷类和萜类为主[21],本研究转录组数据为其生物合成通路及调控提供重要的依据。
转录因子在基因表达上起到重要的调控作用。C2H2、AP2/ERF、bHLH、MYB等家族在植物细胞苯丙烷类、甲羟戊酸代谢途径中起到调控作用[22]。越南安息香Unigenes覆盖了plantTGdb数据库54个转录因子家族,涉及到次生代谢调控的转录因子较多,说明了越南安息香植物体中复杂的转录调控机制。
转录组数据应用于构建遗传图谱和分析遗传多样性是其另外的功用[23],本研究挖掘了越南安息香8 712个Unigenes的10 974个SSRs位点,重复类型以二核苷酸为主,三核苷酸次之,与以二核苷酸为主的当归[23]的研究一致。双核苷酸重复以AG/CT类型为主,三核苷酸重复以AAG/CTT类型为主,与当归[23]、甘草[23]等植物相同。初步认为,植物中SSRs重复以双核苷酸和三核苷酸重复为主,同时,不同的物种又有所区别。
越南安息香的转录组数据研究初步获得了大量的基因信息,后续通过进一步系统分析,以期揭示越南安息香中苯丙素脂、三萜类等活性物质的生物合成、调控机制,以及该物种的遗传特征,为越南安息香资源的开发及利用提供数据基础。
利益冲突 所有作者均声明不存在利益冲突
[1] Burger P, Casale A, Kerdudo A,. New insights in the chemical composition of benzoin balsams [J]., 2016, 210: 613-622.
[2] Orchard A, Viljoen A, van Vuuren S. Antimicrobial essential oil combinations to combat foot odour [J]., 2018, 84(9/10): 662-673.
[3] Shin S. Anti-activities of plant essential oils and their combination effects with ketoconazole or amphotericin B [J]., 2003, 26(5): 389-393.
[4] Min B S, Oh S R, Ahn K S,. Anti-complement activity of norlignans and terpenes from the stem bark of[J]., 2004, 70(12): 1210- 1215.
[5] Min B S, Na M K, Oh S R,. New furofuran and butyrolactone lignans with antioxidant activity from the stem bark of[J]., 2004, 67(12): 1980-1984.
[6] Wang F, Hua H M, Pei Y H,. Triterpenoids from the resin ofand their antiproliferative and differentiation effects in human leukemia HL-60 cells [J]., 2006, 69(5): 807-810.
[7] Sabbah D A, Al-Tarawneh F, Talib W H,. Benzoin schiff bases: Design, synthesis, and biological evaluation as potential antitumor agents [J]., 2018, 14(7): 695-708.
[8] Castel C, Fernandez X, Lizzani-Cuvelier L,. Characterization of the chemical composition of a byproduct fromgum [J]., 2006, 54(23): 8848-8854.
[9] Ha J, Kang Y G, Lee T,. Comprehensive RNA sequencing and co-expression network analysis to complete the biosynthetic pathway of coumestrol, a phytoestrogen [J]., 2019, 9(1): 1934.
[10] Xu Z C, Peters R J, Weirather J,. Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues ofand tanshinone biosynthesis [J]., 2015, 82(6): 951-961.
[11] Grabherr M G, Haas B J, Yassour M,. Full-length transcriptome assembly from RNA-Seq data without a reference genome [J]., 2011, 29(7): 644-652.
[12] Zhang Z H, Cao B L, Li N,. Comparative transcriptome analysis of the regulation of ABA signaling genes in different rootstock grafted tomato seedlings under drought stress [J]., 2019, 166: 103814.
[13] Tang J R, Lu Y C, Gao Z J,. Comparative transcriptome analysis reveals a gene expression profile that contributes to rhizome swelling invar.[J]., 2020, 154(4): 515-523.
[14] Cao Z, Li L, Kapoor K,. Using a transcriptome sequencing approach to explore candidate resistance genes againstin the wild lentil species Lens ervoides [J]., 2019, 19(1): 399.
[15] Mortazavi A, Williams B A, McCue K,. Mapping and quantifying mammalian transcriptomes by RNA-Seq [J]., 2008, 5(7): 621-628.
[16] Haile Z M, Nagpala-De Guzman E G, Moretto M,. Transcriptome profiles of strawberry () fruit interacting withat different ripening stages [J]., 2019, 10: 1131.
[17] Liu Y L, Zhang P F, Song M L,. Transcriptome analysis and development of SSR molecular markers infisch [J]., 2015, 10(11): e0143017.
[18] Wang S Q, Wang B, Hua W P,. De novo assembly and analysis oftranscriptome and identification of genes involved in polysaccharide biosynthesis [J]., 2017, 18(9): E1950.
[19] Zhou Y Q, Wang X N, Wang W S,. De novo transcriptome sequencing-based discovery and expression analyses of verbascoside biosynthesis-associated genes intuberous roots [J]., 2016, 36(10): 1-11.
[20] Mudado M d e A, Ortega J M. A picture of gene sampling/expression in model organisms using ESTs and KOG proteins [J]., 2006, 5(1): 242-253.
[21] Hovaneissian M, Archier P, Mathe C,. Analytical investigation of Styrax and benzoin balsams by HPLC-PAD-fluorimetry and GC-MS [J]., 2008, 19(4): 301-310.
[22] Sun R Z, Cheng G, Li Q,. Light-induced variation in phenolic compounds in cabernet sauvignon grapes (L.) involves extensive transcriptome reprogramming of biosynthetic enzymes, transcription factors, and phytohormonal regulators [J]., 2017, 8: 547.
[23] Chen C, Chen Y J, Huang W J,. Mining of simple sequence repeats (SSRs) loci and development of novel transferability-across EST-SSR markers from de novo transcriptome assembly of[J]., 2019, 14(8): e0221040.
Transcriptiomic data analysis of roots, stems, and leaves of
WANG Xiao-min1, XU Hua-hua2, ZHAN Ruo-ting1, MA Xin-ye1
1. Key Laboratory of Chinese Medicinal Resource from Lingnan, Ministry of Education, Research Center of Chinese Herbal Resource Science and Engineering, South Medicine Research and Development Laboratory of National Engineering Research Center for Chinese Patent Medicine, Guangzhou University of Chinese Medicine, Guangzhou 510006, China 2. School of Chinese Materia Medica, Guangzhou University of Chinese Medicine, Guangzhou 510006, China
To obtain the transcriptome information characteristics of roots, stems, and leaves by transcriptome sequencing of.The roots, stems, and leaves ofwere selected as the research objects, and Illumina HiSeqTM2000 was used to carry out the transcriptome sequencing analysis of these roots, stems, and leaves.A total of 53 835 045 high-quality sequences (clean reads) were obtained from the roots, stems and leaves by transcriptome sequencing, and 69 151 Unigenes were assembled by Trinity de novo, with an average length of 778 nt. BLAST analysis showed that 41 412 (59.89%), 31 189 (45.10%), 25 539 (36.93%), and 16 749 (24.22%) Unigenes were annotated in the Nr, Swiss-port, KOG, and KEGG databases, respectively, which could be classified into 46 branches of cell components, biological processes and molecular functions of the three major classes in GO classification, involving 129 KEGG standard metabolic pathways, of which 31 had secondary metabolic pathways. There were 3 461 protein coding frame sequences, involving 54 families of higher plant transcription factors. MISA software was used to mine 10 974 simple repeat sequences (SSRs), in which the two-base repeat SSRs was the most abundant, with 6 282 (57.24%), and the five-base repeat SSRs were the least, accounting for 2.45%.The transcriptome information characteristics ofroots, stems, and leaves were obtained by high-throughput technology and bioinformatics analysis, which laid a foundation for the study of functional identification, secondary metabolic pathway analysis and regulation mechanism of.
(Pierre) Crail ex Har-tw.; transcriptome; functional genes; metabolism pathway; simple sequence repeats
R282.12
A
0253 - 2670(2021)08 - 2392 - 08
10.7501/j.issn.0253-2670.2021.08.023
2020-08-06
国家自然科学基金青年基金项目(81102764);广东省教育厅重点提升平台建设项目—岭南中药资源教育部重点实验室(2014KTSPT016)
王小敏(1990—),男,硕士研究生,研究方向为芳香药用植物资源学。Tel: 19860209295 E-mail: 807548046@qq.com
马新业(1976—),男,博士,副研究员,主要从事中药资源学研究工作。Tel: 15817036306 E-mail: usermxy@163.com
[责任编辑 时圣明]
