无PAM限制的CRISPR/Cas9系统构建及效率验证
2021-04-21秦怀远李和刚辛京京赵金山
秦怀远,李和刚,张 宁,辛京京,赵金山
(青岛农业大学动物科技学院,山东青岛 266109)
CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats)源于Ⅱ型微生物适应免疫系统,在该系统中细菌会通过入侵噬菌体和质粒来破坏基因组信息保护自己[1]。当病毒侵入个体,会释放出病毒DNA,当细菌检测到病毒DNA 存在时会释放出2 种RNA,其中sgRNA 与病毒DNA 相结合配对,另一种RNA 指导合成Cas9 蛋白并与sgRNA 组成复合物,从而切断病毒DNA 使其失效[2-3]。CRISPR/Cas9 系统需要通过识别在Cas9 靶位点上的一个原间隔子相邻基序(PAM)[4-6]序列进行特异性靶向基因编辑。目前广泛使用的化脓性链球菌SpCas9 识别的PAM 为NGG(N为任意碱基,G 为鸟嘌呤),也就是说它仅仅能识别靶向基因组中1/16 的位点[7],所以NGG 的PAM 并不能满足对广谱基因的编辑需求,因此研究者们试图通过降低PAM 对CRISPR/Cas9 使用的限制,来拓宽CRISPR/Cas9 系统的编辑范围。
Kleinstiver 等[8]在野生型SpCas9 的基础上,进行R1335V/L1111R/D1135V/G1218R/E1219F/A1322R/T1337R 7 个位点的定点突变,成功构建出了PAM 仅为NG的Cas9 突变体SpCas9-NG。Hu 等[9]运用噬菌体辅助持续演化(Phage-Assisted Continuous Evolution,PACE)[10]的方法来进化并拓展其识别的PAM 序列获得了xCas9。xCas9 比SpCas9 有更高的DNA 靶向特异性,而且将PAM 的限制也降至了NG。Liu[11]团队证实xCas9 在兔子中具有扩展PAM 兼容性和增强的碱基编辑效率,可用于生物体中的精确基因修饰。而Chatterjee 等[12]发现一种SpCas9 的直系同源物,来自犬链球菌的ScCas9,其与SpCas9 具有89.2%的同源性,在CRISPR/Cas9 晶体REC3 结构域[13-14]中,于367 至376 位插入了10 个带正电荷的氨基酸(IKHRKRTTKL),并且在1 337 和1 338 位置上插入了另外2 个氨基酸(KQ)[15],最后PAM 进一步放宽至NNGN,而且也有较强的靶向切割活性。这些发现进一步拓展了CRISPR 系统的靶向范围。
虽然上述研究已经将CRISPR/Cas9 的PAM 限制降低至NG(SpCas9-NG、xCas9 等)以及NNG(ScCas9 等),但是想要更加彻底地使用CRISPR/Cas9 进行全基因组的无限制基因编辑,就必须进一步降低PAM 的限制。所以,在本实验中,ScCas9 在有关控制PAM 的结构域中多出12 个氨基酸,在野生型SpCas9 氨基酸序列的基础上,引入ScCas9 的氨基酸序列特点(插入特定的12 个氨基酸),期望可以将野生型的SpCas9 的NGG PAM 降低至ScCas9 的NNG PAM;同时在SpCas9-NG 和xCas9 氨基酸序列的基础上,引入ScCas9 的氨基酸序列特点(插入特定的12 个氨基酸),期望能将SpCas9-NG 或xCas9 的PAM(NGN)限制彻底消除。
1 材料与方法
1.1 实验材料 迪庆绵羊皮肤成纤维细胞(DQSHS1)购自中国科学院昆明动物研究所,目录号:KCB90012S;pX330(编码野生型SpCas9)质粒购自Addgene 公司,货号:#42230,pX330-NG(编码SpCas9-NG)、pX330-xCas9(编码xCas9)质粒由本实验室在pX330基础上构建而成;SSA-DKK2 报告载体由本实验室保存;BbsI 限制性内切酶购自NEB 公司,货号:R3539S;DH5α感受态细胞购自TaKaRa 公司,货号:AIG1025A;T4 DNA 连接酶购自天根生物公司,货号:B0202S;DNA 胶回收试剂盒购自天根生物公司,货号:DP209-02;去内毒素质粒提取试剂盒购自OMEGA 公司,货号:D6948-01;Lipo 3000 脂质体购自美国Invitrogen公司,货号:L3 000015;双荧光素酶报告基因检测试剂盒购自北京原平皓生物科技有限公司,货号:LF103-01;DNA 寡核苷酸由擎科公司合成。
1.2 载体构建
1.2.1 向导RNA 通用表达载体pCas9-sgRNA 设计与构建 向导RNA 通用表达载体pCas9-sgRNA 载体依次包含如下元件:
U6 启动子:
GAGGGCCTATTTCCCATGATTCCTTCATATTTG CATATACGATACAAGGCTGTTAGAGAGATAATTGG AATTAATTTGACTGTAAACACAAAGATATTAGTACA AAATACGTGACGTAGAAAGTAATAATTTCTTGGGT AGTTTGCAGTTTTAAAATTATGTTTTAAAATGGACT ATCATATGCTTACCGTAACTTGAAAGTATTTCGATT TCTTGGCTTTATATATCTTGTGGAAAGGACGAAAC ACC
spacer 克隆位 点(2 个反向 的BbsI 位 点,2 个BbsI 位点之间插入330 bp 随机序列):
GGGTCTTCG
GGCGAGCTGCACGCTGCCGTCCTCGATGTTGT GGCGGATCTTGAAGTTCACCTTGATGCCGTTCTTC TGCTTGTCGGCCATGATATAGACGTTGTGGCTGTT GTAGTTGTACTCCAGCTTGTGCCCCAGGATGTTGC CGTCCTCCTTGAAGTCGATGCCCTTCAGCTCGATG CGGTTCACCAGGGTGTCGCCCTCGAACTTCACCTC GGCGCGGGTCTTGTAGTTGCCGTCGTCCTTGAAGA AGATGGTGCGCTCCTGGACGTAGCCTTCGGGCATG GCGGACTTGAAGAAGTCGTGCTGCTTCATGTGGT CGGGGTAGCGGCTGAAGCA
AGAAGACCT
sgRNA 下游序列:GTTTTAGAGCTAGAAATAGC AAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAA AAAGTGGCACCGAGTCGGTGC
U6 终止子:TTTTTT
bGH polyA:
CTAGAGCTCGCTGATCAGCCTCGACTGTGCCT TCTAGTTGCCAGCCATCTGTTGTTTGCCCCTCCCC CGTGCCTTCCTTGACCCTGGAAGGTGCCACTCCCA CTGTCCTTTCCTAATAAAATGAGGAAATTGCATCG CATTGTCTGAGTAGGTGTCATTCTATTCTGGGGGG TGGGGTGGGGCAGGACAGCAAGGGGGAGGATTG GGAAGAgAATAGCAGGCATGCTGGGGA
把上述序列插入到pUC57 的EcoR V 位点,此过程委托青岛擎科生物技术有限公司完成。
1.2.2 靶标的设计与载体构建 在绵羊基因库中找出绵羊DKK2基因(NC_040257.1,21620923~21621362,440 bp),并将其第一外显子编码复制出来,设计NNG PAM、NNN PAM 的靶标,合成相应的寡核苷酸,其序列信息如表1,引物委托青岛擎科生物技术有限公司合成。
将NNGT1、NNGT2、NNGT3、NNNT1、NNNT2、NNNT3 的上游引物和下游引物两两退火,在PCR 仪中进行退火过程。退火条件设定为95℃ 5 min,72℃ 10 min,后置于冰上,得到6 条双链寡聚核苷酸。
使用BbsI 内切酶对pCas9-sgRNA 通用表达载体在37℃恒温培养箱进行3 h 的酶切,其中酶切体系为50 μL:5 μL Buffer、2 μL 内切酶、28 μL 超纯水,15 μL pCas9-sgRNA。将酶切产物进行凝胶电泳确定切开后,对酶切产物的3 300 bp 条带进行切胶回收实验,将6 条退火的双链寡核苷酸与胶回收产物用T4 连接酶在4℃冰箱进行过夜连接,其中连接体系为10 μL:4 μL 退火引物,4 μL 酶切产物,1 μL T4 连接酶,1 μLBuffer;次日将所得连接产物用DH5α进行转化,涂板,并将培养皿放置于37℃培养箱进行过夜培养。

表1 DNA 寡核苷酸序列
挑取单菌落加入到含有1 mL LB(含50 mg/mL 氨苄青霉素)的1.5 mL 离心管中,37℃ 200 rpm 摇菌7~8 h,分别使用NNGT1、NNGT2、NNGT3、NNNT1、NNNT2、NNNT3 的上游引物与特异性反向引物X2-R 进行菌落PCR 扩 增,其中PCR 反应体系为25 μL:22 μL 金牌mix、1 μL 上游引物、1 μL X2-R、1 μL 菌液。PCR 反应参数为热盖105℃,95℃ 5 min,94℃ 30 s,60℃ 30 s,72℃ 30 s,上述过程循环29 次,72℃ 5 min,16℃保存。检测相应单菌落,扩增后通过凝胶电泳,筛选120 bp 左右条带的阳性菌落送往青岛擎科生物技术有限公司进行测序验证(测序引物为X2-R)。
1.2.3 SpCas9-NNG、ngCas9NNN、xCas9NNN 突变体的构建 在原有的野生型SpCas9、突变体SpCas9-NG 和xCas9 的基础上,进一步进行ScCas9 中的12 个氨基酸序列的插入。野生型SpCas9、突变体SpCas9-NG 和xCas9 对应的表达Cas9 蛋白的氨基酸序列,在367 位置插入IKHRKRTTKL 十肽,在1 337、1 338 2 个位点插入KQ 二肽。此过程委托青岛擎科生物技术有限公司完成。
1.3 细胞转染 取生长状态良好的绵羊成纤维细胞,先将细胞进行稳定培养传代,在3~5 代细胞生长活性较强时进行24 孔板的铺板。在孔内细胞密度达到75%左右时进行细胞瞬时转染。本次实验分SpCas9-NNG、ngCas9NNN 和xCas9NNN 3 个部分,每个实验有4 组,分别为对照组和3 个实验组,3 个实验组分别由pCas9-sgRNA NNNT1、pCas9-sgRNA NNNT2、pCas9-sgRNA NNNT3 与突变体、SSA-DKK2 表达载体进行共转染,将未连接sgRNA 的空白通用表达载体pCas9-sgRNA与突变体、SSA-DKK2 表达载体作为对照组,该实验采用单一变量法,对照组加未连接靶标引物的pCas9-sgRNA 通用表达载体。其他实验组分别加入pCas9-sgRNA T1、pCas9-sgRNA T2、pCas9-sgRNA T3。实验在不同代数的细胞重复转染2 次,转染48 h 后,进行双荧光素酶活性的测定,以达到验证3 种突变体是否具有切割活性的目的。
1.4 双荧光素酶报告基因检测
1.4.1 细胞裂解 将转染48 h 后的24 孔板中的培养基吸出来,每孔加入100 μL PBS 进行润洗,重复2 次,除去死掉的细胞以及杂质。每孔加入100 μL 稀释好的细胞裂解液,常温静置30 min 进行裂解。
1.4.2 萤火虫荧光素酶活性测定 将裂解好的细胞进行柔和吹打混匀,取100 μL Luciferase Assay Reagent 加入Promega GloMax 20/20 发光检测仪测定管底部,加入20 μL 待测样品(裂解好的细胞),轻轻混匀后,放入仪器中进行检测。
1.4.3 海肾荧光素酶活性测定 萤火虫荧光素酶活性测定之后,直接向取出的离心管中加入100 μL 的终止液SR,轻柔吹打混匀后,放入仪器进行检测,Stop Reagent 可以立即终止萤火虫荧光素酶发光,并且同时启动海肾荧光素酶发光反应。记录海肾荧光素酶反应强度(RLU2)与萤火虫荧光素酶反应强度(RLU1)的数值,计算2 组数据的比值,即RLU2/RLU1(简写为R2/R1)。
1.5 统计分析 将双荧光素酶活性检测结果数据RLU1、RLU2 记录下来,并计算R2/R1,记录于Excel 表格中,计算试验组3 组的平均值,然后使用t 检验对各组结果差异显著性进行分析(将试验组与对照组进行比较,试验组之间不做对比),P≤0.01 为差异极显著,P≤0.05为差异显著,P>0.05 为差异不显著。
2 结果与分析
2.1 引物退火 如图1 所示,两条单链寡聚核苷酸链通过退火,配对形成一条20 bp 的双链寡聚核苷酸链,通过凝胶电泳,1~6 个孔均为明亮单一条带,说明引物退火成功。

图1 引物退火结果
2.2 pCas9-sgRNA 酶切如 图2 所示,pCas9-sgRNA 空载体通过BbsI 酶切,产生3 300 bp、300 bp 2 条带,切胶回收3 300 bp 条带。
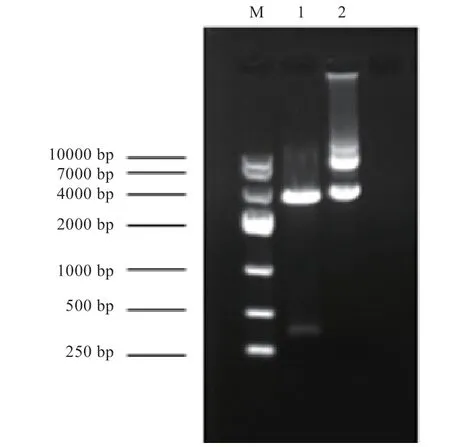
图2 pCas9-sgRNA 酶切结果
2.3 菌液PCR 及测序结果 NNG、NNN 各有3 对退火引物的连接产物,每组挑5 个单个菌落进行菌液PCR,PCR结果通过凝胶电泳检测,结果见图3-A,1~15 为pCas9-sgRNA NNGT1,pCas9-sgRNA NNGT2 以及pCas9-sgRNA NNGT3,其中只有11 显阴性,其余全为阳性,挑选4、5、8、9、12、13 送至青岛擎科生物技术有限公司进行测序。图3-B 中,1~15 为pCas9-sgRNA NNNT1、pCas9-sgRNA NNNT2 以及pCas9-sgRNA NNNT3,其中4、5、7、10、14、15 号孔电泳条带单一明亮,显示为阳性,送至青岛擎科生物技术有限公司进行测序。

图3 菌液PCR 鉴定结果
2.4 pCas9-sgRNA 靶标载体构建结果 如图4 所示,A、B、C、D、E、F 测序峰图表明,靶标均准确连入通用表达载体pCas9-sgRNA 上,NNG、NNN 2 种PAM 的靶标载体pCas9-sgRNAT1、pCas9-sgRNAT2、pCas9-sgRNAT3 构建完成。
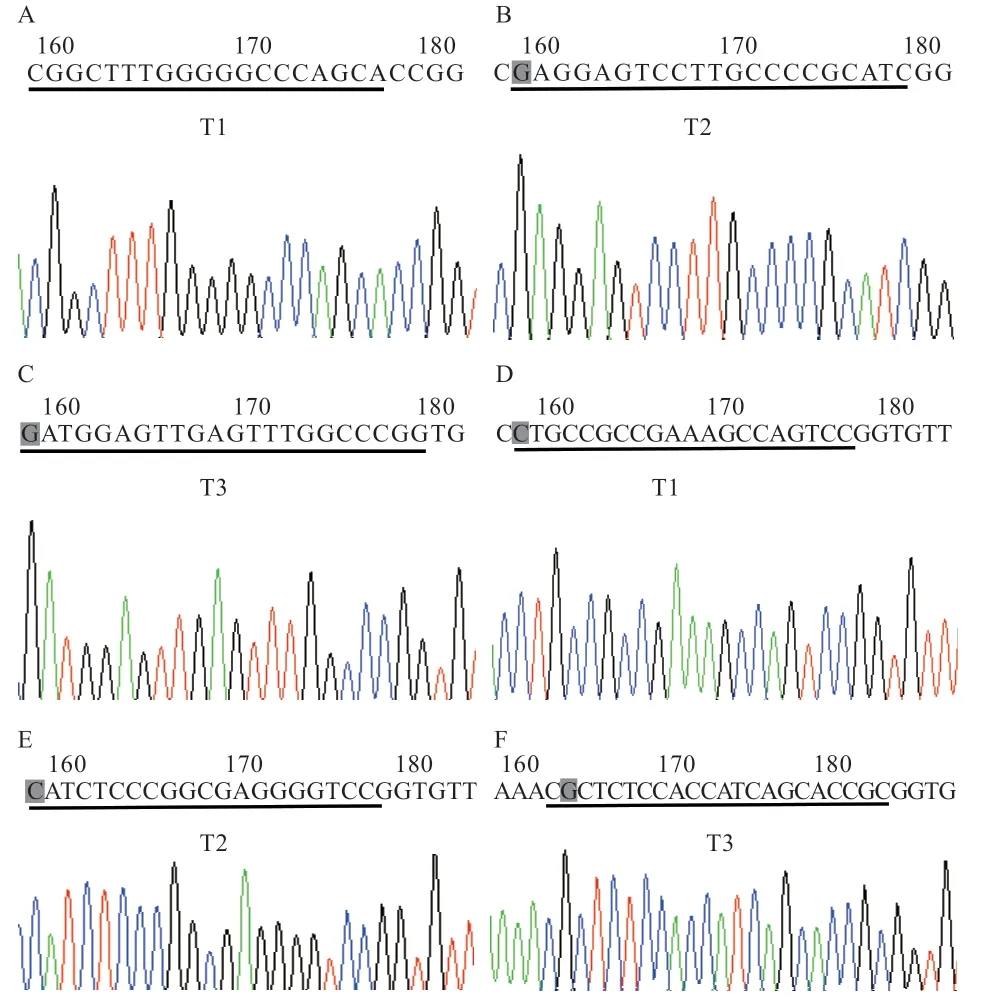
图4 靶标载体测序峰图
2.5 双荧光素酶检测结果 双荧光素酶检测后,将实验组结果与对照组进行SPSS 分析,对照组无sgRNA 进行引导,所以ngCas9NNN 突变体无切割效率,而实验组ngCas9NNN 突变体在sgRNA 的引导下,对目的基因进行了有效的靶向切割,如图5-A 所示,实验组1、2 与对照组相比差异极显著;实验组3 与对照组相比差异显著。结果说明ngCas9NNN 突变体有显著的DNA双链切割活性。
林雪川在广安当地经营了数家公司,涵盖了水业、建筑、物流、旅游开发等多个行业,但效益一直不好,欠债上千万元。
如图5-B 所示,xCas9NNN 突变体试验,与对照组进行的双荧光素酶检测结果对比,只存在一定的切割活性,但活性不高。
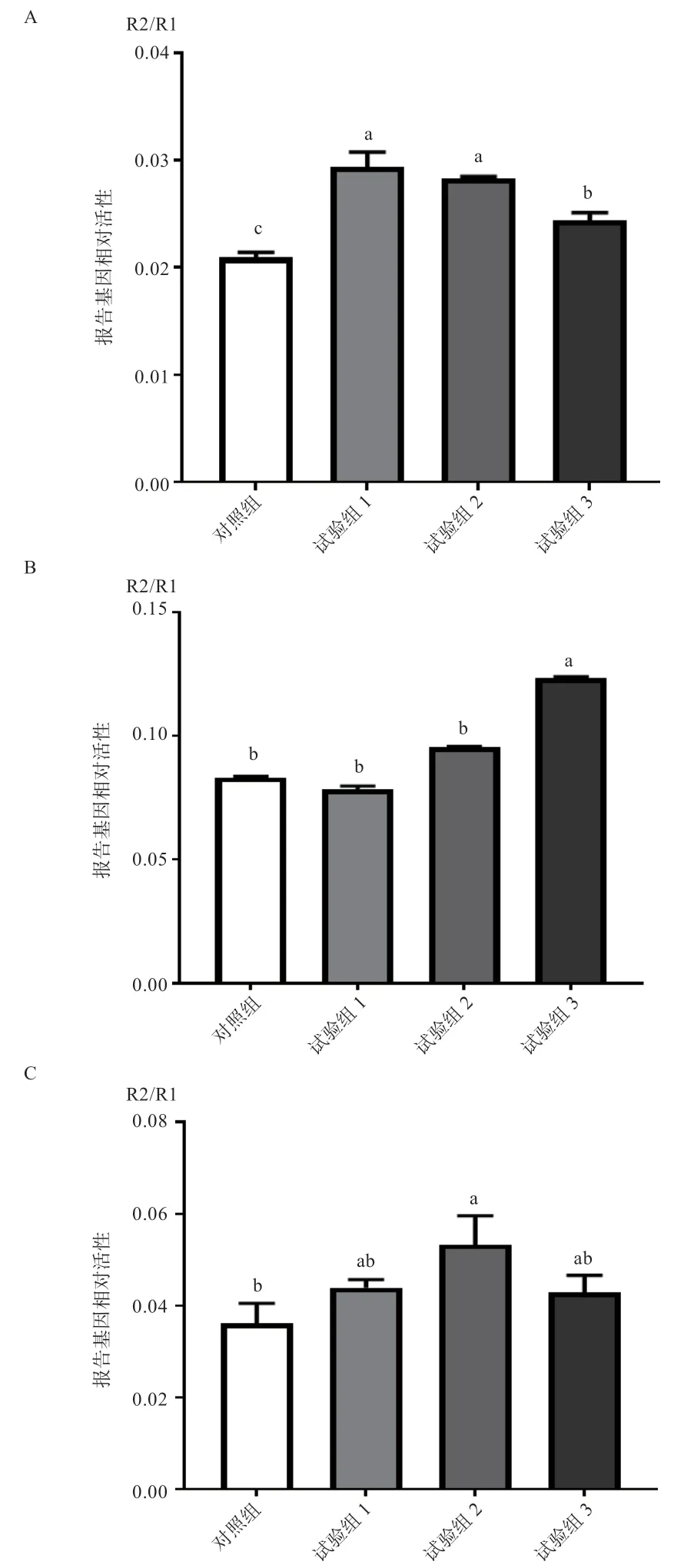
图5 双荧光素酶报告载体检测法检测结果
如图5-C 所示,SpCas9-NNG 突变体试验,与对照组进行的双荧光素酶检测结果对比,有一定切割活性,但是活性不高。
3 讨 论
本研究针对在PAM 识别相关的结构域中,寻找与PAM 识别有关的关键位点,进行氨基酸的突变插入,没有进行定点突变。实验是在ScCas9、野生型SpCas9、xCas9 和SpCas9-NG 的基础 上,将2 种Cas9 蛋白的PAM 限制特点进行中和,优势互补,针对SpCas9-NG和xCas9,在第3 位PAM 的限制蛋白序列中,引入SpCas9-NG 和xCas9 的氨基酸序列特点,在第2 位PAM 的限制蛋白序列中,引入了ScCas9 的第2 碱基的相关蛋白结构域中的氨基酸取代组合,期望进一步扩大野生型SpCas9、xCas9、SpCas9-NG 的编辑范围。
在晶体结构分析中,SpCas9 识别NGG,主要是通过R1333 和R1335 这2 个氨基酸分子结构侧链与NGG PAM 的第2 位和第3 位的核碱基之间的氢键识别[16]。野生型SpCas9 的氨基酸序列中,E1219 位置上的谷氨酸与R1335 的精氨酸,在空间结构会形成一个双齿盐桥[17-18],这个双齿盐桥保持了其对PAM 第3 位核碱基的识别稳定性,所以在xCas9 的氨基酸序列中,E1219的定点突变(E1219V)是xCas9 降低PAM 识别限制的至关重要的突变,它的存在降低了E1219 与R1335的盐桥的空间作用力,使得R1335 在识别PAM 过程中的结构松动,放松对第3 位核碱基G 的识别,从而使xCsa9 的PAM 限制与野生型SpCas9 相比降至NG。而与野生型SpCas9 的氨基酸序列比较,其他6 个位点的突变则是为了使xCas9 的DNA 靶向特异性提高。
本研究中,SpCas9-NNG、ngCas9NNN 和xCas9NNN都是在野生型SpCas9、SpCas9-NG 和xCas9 的氨基酸序列基础上,引入ScCas9 的氨基酸插入特点,最后得到突变体SpCas9-NNG、ngCas9NNN 和xCas9NNN。野生型SpCas9 的PAM 为NGG,只单纯的加入12 个氨基酸,在E1219 位点没有进行任何突变,所以可能不会破坏E1219 位置上的谷氨酸与R1335 的精氨酸之间的盐桥作用力,而且没有使Cas9 蛋白放松对第二位核碱基的识别,猜想可能由于野生型SpCas9 与ScCas9本身存在的差异,致使野生型SpCas9 仅仅通过插入12个氨基酸,并不能降低其对第2 位核碱基识别的放松。对于突变体ngCas9NNN 和xCas9NNN,这两者本身的氨基酸序列也有着差别,其中在关键的1 219 位点上,两者的氨基酸也有所不同,xCas9 在野生型SpCas9 基础上将谷氨酸突变为缬氨酸(E1219V),而SpCas9-NG 则在野生型SpCas9 的基础上将1 219 位的谷氨酸换成苯丙氨酸(E1219F)。而最近Walton 等[19]利用结构导向工程,通过一系列的氨基酸取代组合,从中细化和筛选2 种CRISPR/Cas9 突变体SpG 和SpRY,同样是为了将PAM 的限制降低,SpG 和SpRY 在E1219的位置也都进行了定点突变,他们将谷氨酸换成谷氨酰胺(E1219Q),同样破坏了盐桥的作用力,目的都是为了减弱E1219 位置上的谷氨酸与R1335 的精氨酸之间双齿盐桥的作用力,但是突变的差异有可能反而对xCas9NNN 造成了其他的空间作用效果,而对于突变体SpCas9-NNG 与ngCas9NNN,SpCas9-NNG 本身在1 219位置没有任何突变,保持着原始的盐桥作用力,使其PAM 相关的结构域本身空间结构就比较坚固,空间作用力强,对于PAM 的特异性识别难以放松。所以,SpCas9-NNG 和xCas9NNN 切割活性的降低,是否因为控制PAM 关键结构域的突变差异造成,还有待进一步的实验验证。
而SpG 和SpRY 在原来NGG PAM 的基础上[19],进一步将PAM 的限制降低至NGN 和NRN,甚至在NYN 的位点也表现出了一定的编辑效率。其中除了在E1219 位置的突变特点外,在R1333 上也进行了大量的讨论,R1333Q 是提高SpRY 切割活性的关键位点,这将是下一步提高SpCas9-NNG 和xCas9NNN 切割活性的重要提示。
本次转染实验利用的SSA-DKK2 报告载体在之前的多次实验中都取得了稳定的效果[20],为双荧光素酶检测[21]提供了验证基础。在结果分析中,仅对本次插入设计的SpCas9-NNG、ngCas9NNN 和xCas9NNN 进行了切割活性以及效率的验证,是对无PAM 限制的CRISPR/Cas9 编辑系统的初探,而实验结果也为进一步拓展CRISPR/Cas9 的应用范围提供了理论参考,对于2 种突变体的靶向特异性、脱靶效率及应用范围的验证,还有待进一步探索。
4 结 论
在本实验中,在野生型SpCas9、SpCas9-NG 和xCas9 氨基酸序列中引入了ScCas9 的氨基酸取代特点,最后得到突变体SpCas9-NNG、ngCas9NNN 和xCas9NNN。通过双荧光素酶检测报告得出的结果,ngCas9NNN 可以识别NNN PAM 的目的序列,并且可以进行有效切割。
