头孢地尼中杂质来源分析与合成
2021-04-16王静吴丹刘龙
王静,吴丹,刘龙,3*
(1.泰州医药高新技术产区园区公共平台服务中心,江苏 泰州 225300;2.广州医科大学 药学院,广东 广州 511436;3.天津佰肽特医药科技有限公司,天津 300000)
头孢地尼(Cefdinir,图1),化学名(6R,7R)-7-[[(2-氨基-4-噻唑基)-(肟基)乙酰基]氨基]-3乙烯基-8-氧代-5硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸,是一种优良的第三代口服头孢类抗生素,由日本藤泽药品工业株式会社研制开发,于1991年首次在日本上市,商品名为:Cefzon®。1997年12月经美国食品药品监督管理局批准上市,商品名为:Omnicef®。1999年在韩国上市,2001年获批在我国上市,商品名为:世扶尼®[1-5]。
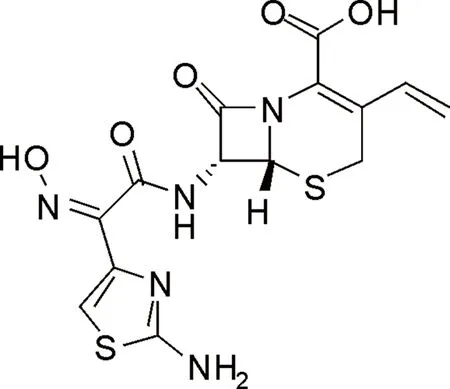
图1 头孢地尼结构Fig.1 Structure of Cefdinir
已经有很多文献报道了头孢地尼原料药的合成[6-18],其中最常用的方法是以7-AVCA(11)和活性酯CAEM(1)为原料,以四氢呋喃和水为溶剂,在三乙胺的作用下缩合得到O-乙酰头孢地尼(12)(以下简称“杂质S”),O-乙酰头孢地尼在氯化铵和碳酸钾的作用下水解得到头孢地尼。《中国药典》2015年版[19]和《美国药典》40版[20]均已收载该品种。《中国药典》2015年版收录的头孢地尼有关物质,对特定杂质A~S进行了定量控制,但没有对《美国药典》40版中的特定杂质噻唑乙酰基甘氨酸肟(2)进行控制。
国内外关于头孢地尼杂质的合成报道较少,部分杂质目前未见文献报道,Prasada Rao等[21]报道了《中国药典》2015年版中的杂质D(8)和杂质G(10)的合成路线,但未提供纯化方法。
因此,本文参考以上两部药典对头孢地尼有关物质的规定,基于头孢地尼的合成工艺路线,对美国药典中规定的特定杂质噻唑乙酰基甘氨酸肟(2)和《中国药典》2015年版中的特定杂质O(7)、杂质D(8)、杂质G(10)和杂质S(12)进行合成、分离和纯化,使其纯度均达到了99%以上,为头孢地尼质量控制中的杂质对照品提供了来源,极大地降低了购买对照品的成本。
1 杂质来源和合成路线
噻唑乙酰基甘氨酸肟(2):化学名N-[(Z)-2-(2-氨基噻唑-4-基)-2-(羟基亚氨基)乙酰基]甘氨酸,由头孢地尼上的β内酰胺环开环裂解产生。本研究以CAEM(1)和Gly为原料,在碱性条件下缩合,酸性条件脱除肟上的乙酰基得到产物(2),产率为30%,纯度达到99.6%,见图2。

图2 噻唑基乙酰基甘氨酸肟合成路线Fig.2 Synthesis of Thiazolylacetyl glycine oxime
杂质O(7):化学名(6R,7R)- 7-[(Z)-2-(2-氨基-4-噻唑基)乙酰氨基]-3-乙烯基-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸,是由头孢地尼起始原料CAEM(1)中的杂质引入并参与后续酰化反应,得到的副产物。以2-(2-氨基噻唑-4-基)乙酸(3)为原料,经过羧基酯化,氨基Trt保护,水解酯基,羧基成酰氯,与7-AVCA(11)缩合,脱Trt保护基制得杂质O(7),产率为11%,纯度99.2%,见图3。
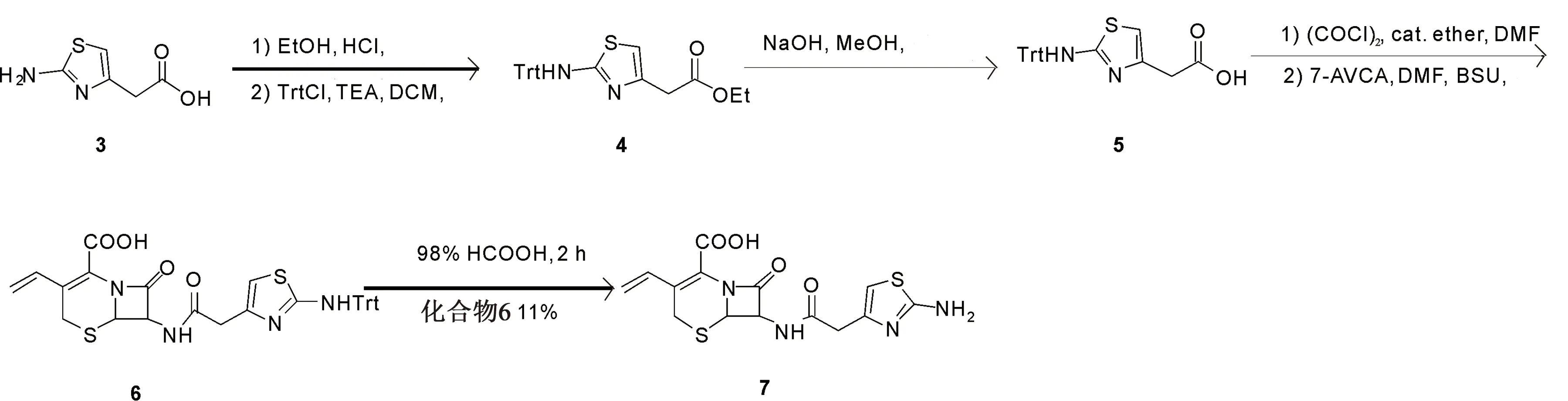
图3 杂质O合成路线Fig.3 Synthesis of impurity O
杂质D(8):化学名(6R,7R)-7-[(Z)-2-(2-氨基-4-噻唑基)-2-(肟基)乙酰氨基]-3-乙烯基-5,8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸,是由头孢地尼氧化得到的杂质。以头孢地尼为起始原料,过氧乙酸氧化得到杂质D(8),产率为27%,纯度99.6%,见图4。
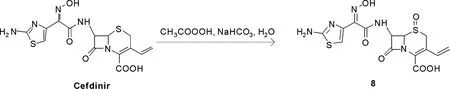
图4 杂质D的合成路线Fig. 4 Synthesis of impurity D
杂质G(10):化学名(6R,7R)-7-[(Z)-2-(2-氨基-4-噻唑基)-2-(肟基)乙酰氨基]-3-甲基-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸,是由原料7-AVCA中的微量杂质,参与后续反应得到的副产物。杂质G(10)与头孢地尼的合成工艺基本一致,即活性酯CAEM(1)与7-ADCA(9)在碱性条件下缩合,再经碱性水解,即可生成,产率为80%,纯度99.1%,见图5。
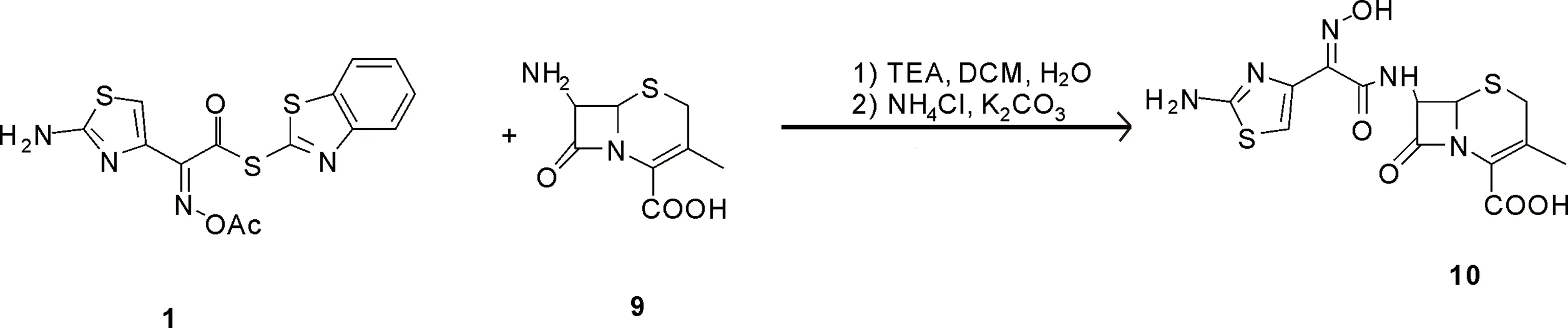
图5 杂质G的合成路线Fig.5 Synthesis of impurity G
杂质S(12):化学名(6R,7R)-7-[(Z)-2-(2-氨基-4-噻唑基)-2-乙酰亚氨基乙酰氨基]-3-乙烯基-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸(25),简称“O-乙酰头孢地尼”。原料7-AVCA(11)和CAEM(1)缩合反应到头孢地尼,当水解不完全时,会生成杂质S。与头孢地尼的合成一致,不同的是酰化反应完成后,不经水解,立即加酸将pH调至6,析出固体,过滤,即得产物,产率80.3%,纯度99.3%,见图6。

图6 杂质S的合成路线Fig.6 Synthesis of impurity S
2 实验部分
2.1 仪器材料
核磁共振氢谱(1H NMR)用BurkerAV-300型核磁共振仪测定,TMS为内标;质谱(MS)用Agilent 1100 ESI-MS型质谱仪测定;红外光谱(IR)用Shimadzu FTIR-8400型红外光谱仪测定,固体KBr压片;熔点仪用天津市分析仪器厂RY-1G型熔点仪测定;液相色谱仪为Shimadzu LC-20A系列检测;薄层色谱(TLC)采用GF254薄层层析硅胶(青岛海洋化工厂);柱层析采用硅胶H(青岛海洋化工厂);合成实验所用溶剂及试剂均为化学纯或分析纯;高效液相色谱法(HPLC)所用试剂均为色谱纯。
2.2 化学合成
噻唑基乙酰基甘氨酸肟:将CAEM (5 g,13 mmol)、甘氨酸(1.95 g,26 mmol)、四氢呋喃(75 mL)、水(75 mL)加入到反应瓶中,室温下搅拌30 min。缓慢滴加三乙胺至反应液pH为8, 滴毕,室温反应36 h。体积分数10%硫酸水溶液调pH至6,析出白色固体,冰浴条件下继续搅拌1 h,析出晶体,抽滤,冷水洗涤。40 ℃真空干燥得白色粉末,二氯甲烷打浆,过滤,得类白色粉末(0.98 g,产率30%,HPLC纯度99.6%), m.p.190~194 ℃。1H NMR (300 MHz, DMSO-d6) ,δ: 1.84(s, 1H), 3.84(d,J=4.8 Hz, 2H), 6.89(s, 1H), 7.13(s, 2H), 8.73(s, 1H), 11.22(brs, 1H); ESI-MS(m/z): 245 [M+H]+; IR (KBr, cm-1):3355, 3303, 3182, 2360, 2341, 1637, 1508, 1482, 1428, 1386, 1354, 1308, 1270, 1105, 1074, 1034, 1013, 954, 848, 749, 624, 541。
杂质O:将化合物3(5 g,32 mmol)、乙醇(88 mL)加入反应瓶中,搅拌制成混悬液,滴加浓盐酸(25 mL),滴毕,加热回流,TLC跟踪至反应完全。降至室温,反应液浓缩至40 mL,加饱和碳酸钠水溶液(37 mL),析出固体,过滤,水洗滤饼(30 mL×2),40 ℃真空干燥,得白色固体。将干燥的白色固体、三苯基氯甲烷(12.1 g,48 mmol)、二氯甲烷(200 mL)加入到反应瓶中,室温搅拌30 min,滴加TEA(4.86 g,48 mmol),继续搅拌15 h,TLC跟踪至反应完全。加水(75 mL)淬灭反应,静置分层,水层用二氯甲烷萃取(60 mL ×2),合并有机层,减压浓缩,得粗品。硅胶柱层析分析(V(石油醚)∶V(乙酸乙酯)=3∶1),得到化合物4。
化合物4 加入反应瓶,在反应瓶中加入3 mol/L氢氧化钠甲醇溶液(20 mL),室温搅拌1 h,TLC监测反应结束。反应液用体积分数10%硫酸水溶液调pH至3.0,析出灰白色固体,过滤,滤饼水洗,40 ℃真空干燥12 h,得化合物5(5.8 g)。
在反应瓶中加入乙醚(10 mL),催化量 DMF,冰浴条件下滴加草酰氯(2.2 g,17.4 mmol),有气体生成,5 min后生成白色固体。反应液减压浓缩,除去乙醚,加无水二氯甲烷(10 mL),搅拌得混悬液。反应液降温至-10 ℃,加入化合物5(5.8 g,14.5 mmol),TLC跟踪至反应完全,得到酰氯化合物。将7-AVCA (3.3 g,14.5 mmol)、DMF(10 mL)、六甲基二硅脲(3.3 g,16 mmol),吡啶(2.5 mL,31 mmol)依次加入到反应液中,反应液维持在-5~-10 ℃搅拌30 min。加二氯甲烷(15 mL),水(15 mL),室温搅拌15 min,静置分层,水层用二氯甲烷(15 mL×2)萃取,合并有机层。有机层浓缩至15 mL,加入乙酸乙酯(18 mL),降温,析出固体,冰箱冷冻过夜,过滤,得白色固体6(4.1 g)。
98%甲酸(10 mL)加入至反应瓶中,冰浴降温至0 ℃,加入化合物6,搅拌至反应液变澄清,升温至室温,继续搅拌2 h,三苯甲基水解,过滤,得到滤饼。滤饼继续用甲酸溶解,得到澄清液体,室温缓慢加入丙酮,冰箱冷冻过夜析晶,过滤,得到滤饼,35 ℃真空干燥,得白色固体杂质O(1.26 g,产率11%,HPLC纯度98.6%),m.p.178~180 ℃(decomp) 。1H NMR (300 MHz, DMSO-d6),δ:3.38(s,2H), 3.57&3.86(ABq,J=17.8 Hz,2H), 5.14(d,J=4.8 Hz,1H),5.31(d,J=11.3 Hz,1H),5.59(d,J=17.6 Hz , 1H),5.69(dd,J=4.8 Hz、8.4 Hz,1H), 6.25(s,1H), 6.86~6.95(m, 2H), 6.90(dd,J=11.3 Hz、17.6 Hz, 1H) ,8.93(d,J=8.4 Hz,1H); ESI-MS(m/z):367[M+H]+; IR(KBr,cm-1):3288, 1764,1657, 1543, 1389, 1346, 1237, 1167, 1119, 711, 556。
杂质D:头孢地尼(3 g,0.007 6 mol)、水(10 mL)加入至反应瓶中,搅拌30 min制成混悬液,温度控制在10~15 ℃,加碳酸氢钠(0.96 g,0.011 mol),反应液搅拌至澄清。降温至0 ℃缓慢滴加30%过氧乙酸(2.1 g,0.008 4 mol),维持0 ℃搅拌2 h,体积分数10%硫酸水溶液调pH至6,搅拌1 h析晶,过滤,水洗,得灰白色固体杂质D(0.84 g,产率27%,HPLC纯度99.6%),m.p.178~183 ℃(decomp)。1H NMR(300 MHz, DMSO-d6), δ:3.45&4.18(ABq,J=18.0 Hz, 2H), 4.94(d,J=4.0 Hz, 1H), 5.23(d,J=11.3 Hz,1H), 5.49(d,J=17.4 Hz , 1H),5.87(dd,J=4.6 Hz,8.3 Hz , 1H), 6.71(s,1H), 6.97(dd,J=11.3 Hz, 17.4 Hz, 1H), 7.24(brs, 2H), 8.49(d,J=8.3 Hz, 1H), 11.6(brs, 1H); ESI-MS(m/z):412[M+H]+; IR(KBr,cm-1):3629, 3398, 2361, 2343, 1774, 1700, 1654, 1637, 1522, 1396, 1121, 1020。
杂质G:反应瓶中依次加入CAEM(2 g,0.009 mol)、7-ADCA(3.75 g,0.010 mol)、水(10 mL)、THF(20 mL),搅拌30 min,滴加TEA(1.0 g,0.01 mol)至pH为8,室温搅拌4 h,溶液变澄清。加二氯甲烷(20 mL),水(10 mL),继续搅拌10 min,静置分层,二氯甲烷层用水萃取(10 mL×2),合并水层。水层加氯化铵(1.3 g,0.025 mol),滴加碳酸钾水溶液(0.9 mol/L)维持pH至8.0~8.5,搅拌2 h,用体积分数10%硫酸水溶液调pH至5,降温至0 ℃,固体析出,抽滤,得到滤饼, 35 ℃真空干燥,得白色固体(3-甲基头孢地尼:杂质G)(2.75 g,收率80%,HPLC纯度99.0%), m.p.179~185 ℃(decomp)。1H NMR (300 MHz , DMSO-d6),δ:2.00(s, 3H, CH3), 3.34&3.54(ABq,J=18.1 Hz,2H),5.09(d,J=4.6 Hz, 1H,),5.70(dd,J=4.6 Hz,8.0 Hz, 1H), 6.69(s, 1H), 7.41(brs, 2H), 9.50(d,J=8.0 Hz, 1H), 11.48(brs, 1H); ESI-MS(m/z): 384[M+H]+; IR(KBr, cm-1): 3274, 3112, 2961, 1762, 1637, 1528, 1378, 1320, 1245, 1189, 1076, 1035, 1013, 750, 605。
杂质S:纯化水(12 mL)、四氢呋喃(36 mL),7-AVCA(3 g,0.013 mol),CAEM(5.9 g,0.016 mol)加入反应瓶,缓慢滴加三乙胺(1.6 g,0.016 mol),使反应液pH在7.5~8.0,滴毕,室温下搅拌4 h(液相监控7-AVCA≤1%)。反应液中加入二氯甲烷(30 mL)、纯化水(30 mL),搅拌10 min,静置分层。二氯甲烷层用纯化水(10 mL×3)萃取,合并水层。水层立即用体积分数10%硫酸水溶液调pH至3,过滤,水洗,35 ℃真空干燥,得灰白色固体杂质S(4.21 g,产率80.3%,HPLC纯度97.5%), m.p.165~170 ℃(decomp)。1H NMR(300 MHz, Acetone-d6),δ:2.2(s, 3H), 3.72&3.92(ABq,J=17.8 Hz,2H),5.33(d,J=3.2 Hz, 1H), 5.36(d,J=3.1 Hz, 1H), 5.64(d,J=17.6 Hz , 1H), 6.03(dd,J=4.8 Hz、8.6 Hz,1H),7.14(m,4H),8.84(d,J=8.6 Hz, 1H); ESI-MS(m/z): 460[M+Na]+; IR(KBr, cm-1): 3434, 3269, 3095, 2969, 1766, 1655, 1628, 1584, 1536, 1370, 1212, 1181, 1001, 918, 852。
3 结果与讨论
本研究成功制备了头孢地尼原料药成品中存在的5个关键杂质,合成路线短,操作简单。其中杂质噻唑乙酰基甘氨酸肟是采用CAEM和Gly缩合得到。杂质O以2-(2-氨基噻唑-4-基)乙酸与7-AVCA缩合再脱Trt保护基得到。杂质D是由头孢地尼直接通过过氧乙酸氧化得到。杂质G是由CAEM与7-ADCA缩合得到。杂质S采用与与头孢地尼的合成方法一致,不同的是酰化反应完成后,不经水解,立即加酸将pH调至6,析出固体得到。5种杂质均经1H NMR和MS确证了结构,纯度经HPLC测定均达到了99%以上。这些杂质的合成,为头孢地尼的质量控制提供了依据和参考,对改进头孢地尼合成工艺,提升原料药质量具有实际意义。
