花生粕中黄曲霉毒素B1测定的方法改进与不确定度评定
2021-04-13杜宇峰刘冠男
杜宇峰,汪 浩,刘冠男
(青岛市产品质量监督检验研究院,山东 青岛 266101)
花生在我国广泛种植,属于我国六大油料作物中的一种[1]。花生经压榨等方式制油后产生的副产品叫做花生粕。花生粕中的蛋白质含量较高且氨基酸种类丰富,被广泛用于饲料中[2]。但是,花生粕在贮存过程中极易发生霉变而产生黄曲霉毒素,主要是黄曲霉毒素B1[3]。黄曲霉毒素B1具有较强的毒性和危害性,有致癌的危险,而且可以导致动植物细胞染色体的畸变[3]。所以,对于花生粕中黄曲霉毒素B1含量的测定和控制具有重要意义。本研究按照GB/T 36858—2018《饲料中黄曲霉毒素B1的测定 高效液相色谱法》对花生粕中黄曲霉毒素B1进行测定,同时进行了不确定度分析,全面地考虑了测定过程中的多个影响因素,并对其进行分析与合成,发现其中对测定结果影响最大的一项要素,提高了测定的准确度和数据的可靠性,以期对测定结果进行正确的评价与规范使用。
1 检测试验
1.1 仪器与设备
ML 204 T/02 型电子天平(量程为0~210 g,感量为0.000 1 g),梅特勒(上海)有限公司产品;1260 型高效液相色谱仪,安捷伦科技有限公司产品;MP 21001型电子天平(量程为0~210 0 g,感量为0.01 g),上海舜宇恒平科学仪器有限公司产品;可调移液器(量程为1~10 mL),宝予德(中国)有限公司产品。
1.2 样品与试剂
样品:花生粕,从青岛市李沧区农贸市场购买,经检验无霉菌污染。
试剂:黄曲霉毒素B1标准溶液(100.6 μg·mL-1,不确定度为1.4 μg·mL-1);甲醇(色谱纯)、乙腈(色谱纯),霍尼韦尔公司产品;三氯甲烷(色谱纯),默克化工技术(上海)有限公司产品;三氟乙酸(TFA)(色谱纯),阿拉丁试剂(上海)有限公司产品;冰乙酸(优级纯),国药集团化学试剂有限公司产品。
1.3 检测方法
依据标准GB/T 36858—2018《饲料中黄曲霉毒素B1的测定 高效液相色谱法》对花生粕中的黄曲霉毒素B1含量进行测定。但为了方便样品的取用以及简化检测流程,在保证样品均一性和稳定性的前提下,对样品制备的方式进行了部分改进。
1.3.1 样品处理
称取花生粕样品1 kg,高速粉碎后过2 mm 孔径试验筛,取孔径2 mm 以下的粉末样品,混合均匀作为待测试样[4]。称取待测试样5.00 g于100 mL锥形瓶中,注入黄曲霉毒素B1提取液(乙腈∶水=84∶16)25.0 mL,以振荡的方式提取黄曲霉毒素B1,提取条件为:转速200 r·min-1,时长60 min。提取完毕将提取液经中速滤纸过滤,然后取10.0 mL 过滤完成后的提取液,用等体积的三氯甲烷进行萃取,进行1 min 的涡旋,混合均匀后静置至清晰分层,将下层的萃取液放入15 mL具塞离心管中,在50 ℃水浴条件下通过氮气吹干,加入200 μL 乙腈水溶液(乙腈∶水=90∶10),复溶后加入700 μL衍生溶液(三氟乙酸∶水∶冰乙酸=20∶70∶10,现用现配),混合均匀后衍生75 min,衍生条件为40 ℃水浴加热,衍生完毕后用0.22 μm微孔滤膜过滤,制备待测试样样液。
1.3.2 标准溶液配制及处理
取100.6 μg·mL-1黄曲霉毒素B1标准溶液1.0 mL,用乙腈稀释至10 mL,制得浓度为10.06 μg·mL-1的中间液,再用乙腈作为溶剂将此中间液稀释100倍制得浓度为100.6 ng·mL-1的标准工作液。使用前再将此标准工作液配制成1、2、5、10、20、50、100 ng·mL-1的标准系列工作液。之后分别吸取0.9 mL的各个标准系列工作液至15 mL具塞离心管中,在50 ℃水浴条件下通过氮气吹干,加入200 μL 乙腈水溶液(乙腈∶水=90∶10),复溶后加入700 μL衍生溶液(三氟乙酸∶水∶冰乙酸=20∶70∶10,现用现配),混合均匀后衍生75 min,衍生条件为40 ℃水浴加热,衍生完毕后用0.22 μm微孔滤膜过滤,制备待测试样样液。
1.3.3 色谱条件
C18色谱柱(250 mm×4.6 mm×5 μm)的流动相为甲醇∶乙腈∶水=2∶1∶7,流速为1 mL·min-1,激发波长为365 nm,发射波长为440 nm,柱温为30 ℃,进样量为20 μL。
1.3.4 计算模型
按以下公式计算黄曲霉毒素B1含量w。
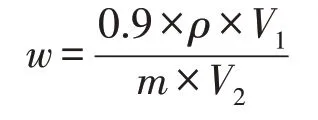
式中:w—样品中黄曲霉毒素B1含量(μg·kg-1),ρ—样品的衍生液在标准曲线上对应的黄曲霉毒素B1含量(ng·mL-1),V1—提取液总体积(mL),V2—用于萃取的提取液体积(mL),m—样品质量(g)。
1.4 试验方法的优化
按照GB/T 36858—2018《饲料中黄曲霉毒素B1的测定 高效液相色谱法》所采取的制样方法,样品的制备过程可简述为:采集至少4 kg的样品,进行缩分至2 kg 后再均匀分为4 份,取其中1 份进行粉碎和过筛后予以检验。此方式需要的样品量较大,且需要进行多次分样后进行样品的粉碎过筛,试验过程较为繁琐,笔者参考GB 5009.22—2016《食品安全国家标准 食品中黄曲霉毒素B 族和G族的测定》[4]中所述的样品制备方法,即称取样品1 kg,粉碎过筛混合均匀后作为待测样品。本研究针对试验用花生粕样品采用两种取样方法进行检测试验,方法1:按照原标准的样品制备方式,取4 kg 样品,缩分至2 kg 后均匀分为4 份,将1 份样品粉碎后过筛,将制备完毕的样品平铺,之后随机对7个部位的样品进行取样,每个部位各取5.00 g,按1.3.1所述检验步骤进行试验。方法2:称取样品1 kg,粉碎过筛混合均匀后作为待测样品,将此待测样品平铺后,随机抽取7 个部位的样品各5.00 g,按照1.3.1所述的检验步骤进行检验。试验结果见表1。
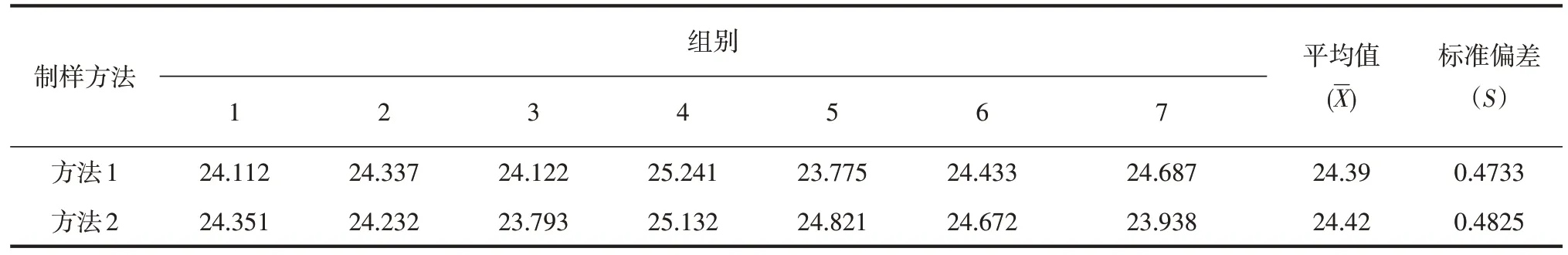
表1 两种制样方法的检测试验结果 ng·g-1
对于两种制样方法的试验结果进行F检验,判断两种方法制备得到的样品黄曲霉毒素B1含量是否存在差异。
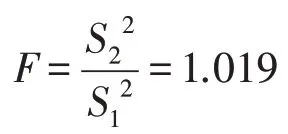
式中:F—两组数据的方差比值;S1—方法1中7次试验数据的标准偏差;S2—方法2中7次试验数据的标准偏差。
因为S2>S1,则有以S22的作为分子进行比较。查F分布表,取显著性水平α=0.01,自由度ƒ1=7,ƒ2=7时,Fs=6.993>F。
说明两组数据的精密度没有显著性差异,所以合并标准偏差为:
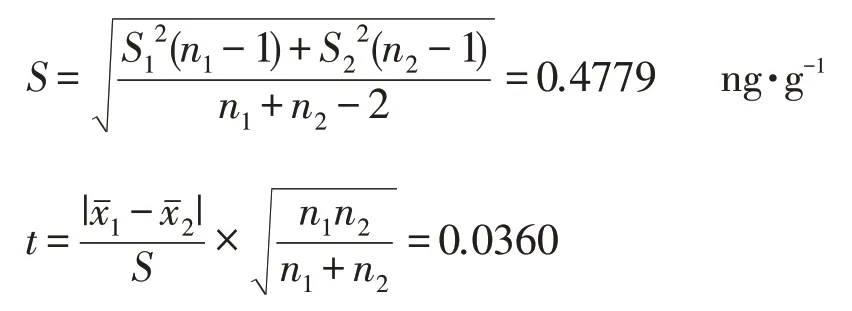
当自由度ƒ=n1+n2-2=12,显著性水平α=0.01时,查t分布表得到t0.01,12=2.681>t。因参照的t、F分布表提供的数值均为单边值,本试验讨论的制样方法之间的差别属于双侧检验的范畴,所以显著性水平应为单侧检验时的2 倍,此时可以得出结论:置信区域为98%的条件下,可以认为两种制样方法得到的样品的黄曲霉毒素B1的含量没有显著性差异。
综上得出结论:两种取样方法在98%的置信条件下无显著性差异。在实际检测中,可以考虑以方法2所述的制样方法对标准既定的取样方法予以优化,以期简化流程、减少样品采集的总量。
2 不确定度来源分析
对于该检测试验的不确定度来源进行分析,因为各个仪器使用温度和环境温度在试验中基本保持恒定,所以由温度导致的不确定度也可以忽略;又因为1 kg样品全部用于制样,且分别进行7次随机取样进行平行测定,所以由样品均匀程度引入的不确定度可并入重复性测定的因素予以讨论。此时,该试验的不确定度来源主要有以下几个方面:1)样品称量;2)标准溶液配制;3)样液的稀释和取用;4)重复性测定。
3 不确定度的分量评定
3.1 样品称量引入的不确定度
试验中使用的是精度为0.000 1 g 的分析天平,从该天平检定证书得到,此天平测量范围在0~50 g时允许误差最大为±0.000 5 g,不确定度按照B 类评定,按照矩形分布分析,取k= 3,从而得到因样品称量而产生的相对不确定度Urel(m)为:
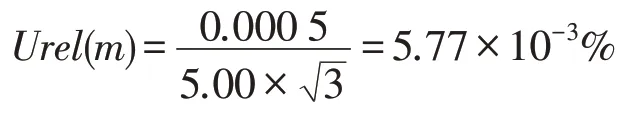
3.2 标准溶液配制产生的不确定度
3.2.1 标准物质的不确定度
本试验使用的标准物质(标准溶液储备液)为黄曲霉毒素B1标准溶液(100.6 μg·mL-1,不确定度为1.4 μg·mL-1),在置信水平95%的条件下,按照B类评定得到,标准物质的不确定度为Urel(C1):
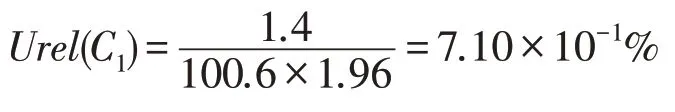
3.2.2 因第1次稀释导致的不确定度
第1次稀释中使用的移液器的量程为1 mL,当吸取液体的体积为1 mL 时,依据JJG 646—2006《移液器检定规程》及移液枪使用说明,此时移液器的允许误差为10 μL,按照矩形分布取得到第1次稀释过程中移液器使用导致的不确定度Urel(C2.1)为:
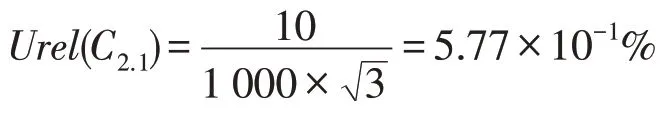
第1 次稀释中使用的10 mL 容量瓶,依据JJG 196—2006《常用玻璃量器》中的检定要求,体积容量误差为±0.020 mL,得到第1 次稀释过程中容量瓶使用导致的不确定度为Urel(C2.2):
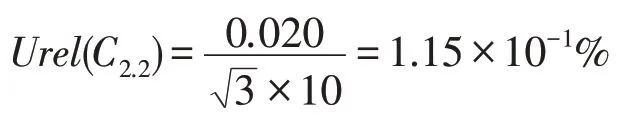
3.2.3 因第2次稀释导致的不确定度
在第2 次稀释过程中,使用的依然为3.2.2 中所述的1 mL 移液器,故得到第2 次稀释中移液器使用导致的不确定度Urel(C3.1)为:
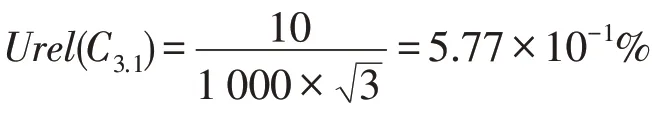
在此过程中,使用的容量瓶规格为100 mL,按照JJG 196—2006《常用玻璃量器》中规定的设备检定要求,此规格容量瓶的体积容量误差为±0.01 mL,可以得到第2次稀释导致容量瓶的使用导致的不确定度Urel(C3.2)为:
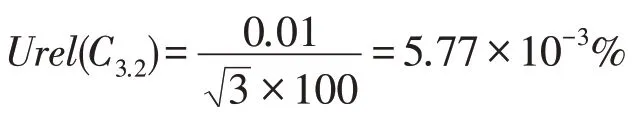
合成以上不确定度分量,则得到本试验使用的黄曲霉毒素B1标准工作液的相对不确定度为。
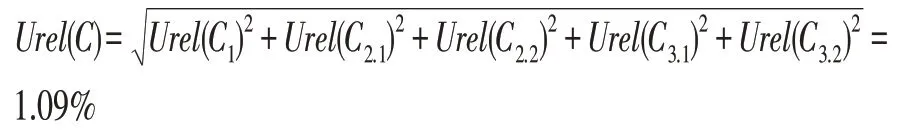
3.3 样液稀释和取用过程中引入的不确定度
提取液总体积为25 mL,提取液通过25 mL 大肚移液管(即A 级单标线吸量管)加入,依据GB/T 12808—2015《实验室玻璃仪器 单标线吸量管》针对规格为25 mL 的A 级单标线吸量管的检定要求,此移液管的容量允许误差为0.030 mL,按照矩形分布取则得到由加入的提取液稀释带入的相对不确定度Urel(V1)为:
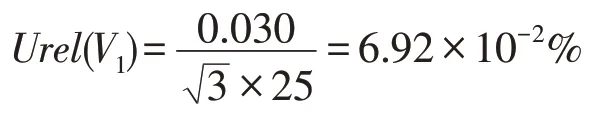
之后吸取提取液10 mL进行试验,采用的移液设备为10 mL移液器,依据JJG 646—2006《移液器检定规程》及该移液枪使用说明得到规格为10 mL的移液器,在取用液体体积为10 mL时的容量允许误差为0.6%,从而得到取用样液导致的相对不确定度Urel(V2)为:
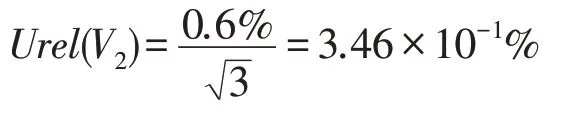
合成以上不确定度得到在样液稀释和取用过程中的相对不确定度Urel(V)为:
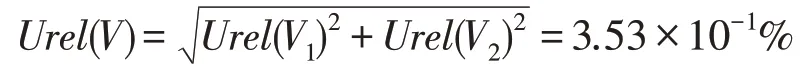
3.4 重复性测定引入的不确定度
针对同一花生粕样品进行7次平行试验,得到的试验结果见表2。
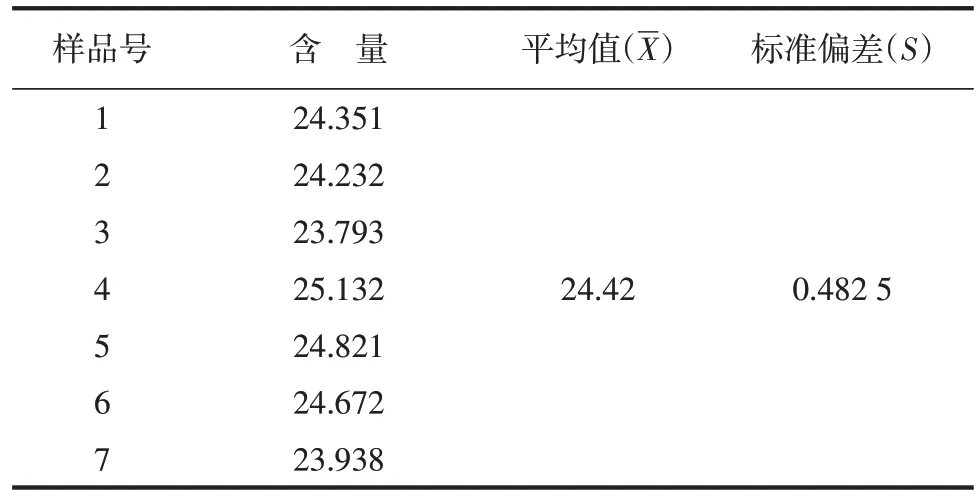
表2 重复性测定结果 ng·g-1
则含量平均值的相对标准不确定度为:

4 不确定度的合成
依据不确定度的合成公式,分别列出四项影响因素对于不确定度的贡献及量值,结果见表3。

表3 花生粕中黄曲霉毒素B1含量测定的不确定度分量
从而得到,合成相对不确定度如下:
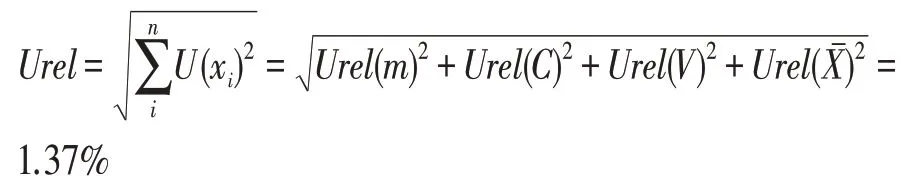
该试验中,花生粕中黄曲霉毒素B1的含量为24.42 ng·g-1,其标准不确定度为:
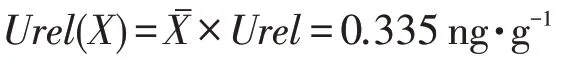
在没有特殊要求的情况下,扩展因子k的取值定为k=2,置信概率P=95%,对黄曲霉毒素B1检测试验结果所得到的标准不确定度进行扩展,得到扩展不确定度Uprel为:
Uprel=k×Urel(X)=0.67 ng·g-1
5 讨 论
本试验对于制样方法的优化,针对试验用的花生粕样品进行7个平行验证,验证得到结论:针对此样品,制样方法可以通过步骤:称取样品1 kg,粉碎过筛,混合均匀后作为待测样品予以简化,对于其他类型的饲料基质还需要比对试验予以验证。
6 结 论
依据标准GB/T 36858—2018《饲料中黄曲霉毒素B1的测定 高效液相色谱法》[5]对于花生粕中黄曲霉毒素B1含量进行测定的过程中,共有以下四个方面的因素对测量不确定度的产生有所贡献,分别为样品称量、样液稀释和取用、重复性测定以及标准溶液配制。由表3不确定度分量得到,通过该方法测定黄曲霉毒素B1含量,影响不确定度的最主要因素为标准溶液配制,样品重复性测定以及样液的稀释与取用对此试验的不确定度引入的贡献适中,样品称量对于试验结果的不确定度贡献最小,最终试验结果的不确定度可以表示为24.42±0.67 ng·g-1。
