基因芯片筛选原发性膀胱癌差异表达基因
2021-04-12史轶良王烨金鹏王行环
史轶良,王烨金鹏,王行环
(武汉大学中南医院泌尿外科,湖北武汉 430071)
膀胱癌(bladder cancer,BC)是世界上第9大常见肿瘤,是男性第4大常见肿瘤,男性患者的中位诊断年龄大约在69岁,其死亡率位居所有肿瘤的第13位,是威胁人类健康的重要疾病[1]。大约90%的膀胱癌为尿路上皮癌。近30年来膀胱癌患者的存活率没有显著变化[2-5]。在发达国家,由于缺乏治疗膀胱癌的有效手段,其威胁程度甚至可以高于其他一些肿瘤。膀胱癌的发病机制涉及多个基因表达通路,而近年基因芯片的大量应用为研究膀胱癌的发生发展机制提供了许多新的方法与思路[6]。本研究通过生物信息学方法对基因芯片GSE13507进行研究,对其包含的膀胱癌与正常外观膀胱癌周围组织样本数据进行分析,为研究膀胱癌的发生发展机制提供可能的方向。
1 材料与方法
1.1 实验材料从GEO数据库中下载编号为GSE13507的基因芯片,该芯片包含256个样本。其中正常膀胱黏膜10例、正常外观膀胱癌周围组织的膀胱黏膜58例、原发膀胱癌165例、复发性非肌层浸润膀胱肿瘤23例。本研究选取原发性膀胱癌与正常外观膀胱癌周围组织的膀胱黏膜作为对照组进行基因微阵列研究[6]。该芯片的平台信息为:GPL6102(Illumina human-6 v2.0 expression beadchip)。芯片的探针注释信息来自Affymetrix公司。原始数据的CEL文件来自pubmed GEO数据库(https://www.ncbi.nlm.nih.gov/gds/)。
1.2 处理方式
1.2.1数据预处理和聚类分析 利用GEOquery包获取原始基因芯片数据并导入R软件,分析确认数据来源的正确性,利用软件获取基因的表达矩阵。对原始基因表达矩阵进行排序,将多次出现的序列取平均值后合并为一项,避免重复计算。通过Affymetrix注释文件获取样本基因的注释信息,并通过基因探针名将其与基因数据对应。用Affy包对其进行RMA背景校正计算,利用样本间的Person相关系数计算,对获取的样本信息进行聚类分析,分析明显错误的芯片数据,对离群样本进行剔除。
1.2.2筛选差异基因 使用Limma数据包进行差异表达分析,设定差异基因的筛选标准为:P<0.05,基因差异倍数(fold change,FC)变化≥1.5或≤-1.5。
1.2.3GO功能富集分析与KEGG通路富集 利用R软件对获取的差异基因表达矩阵进行GO富集分析。使用标准化P值进行统计学意义判断,P<0.05则认为差异有统计学意义。同时进行通路富集分析,检测差异基因表达所在的通路。
1.2.4使用数据库与cytoscape分析蛋白相互作用网络 蛋白互作数据库可以了解各个基因所表达的蛋白之间的相互作用关系。将筛选所得的差异基因利用数据库进行综合分析,获得蛋白互作网络,设置阈值评分为>0.4分,将结果导出。
1.2.5筛选核心蛋白 将前述结果导入cytoscape,并利用其网络分析功能计算差异基因所表达蛋白相互作用的密度与广度。根据蛋白作用的密集程度可以筛选出网络中心节点,其对应的蛋白即为核心作用蛋白。
1.2.6核心蛋白验证 GEPIA数据库包含多种肿瘤数据,利用其膀胱癌数据对所获得的核心蛋白进行综合分析,并验证其功能。
2 结 果
2.1 聚类分析结果对样本数据进行聚类分析显示,原发膀胱癌165例(实验组)与正常外观膀胱癌周围组织的膀胱黏膜58例(对照组)聚类良好,对数据进行归一化处理,用热图的方式进行可视化,红色代表上调的基因,蓝色代表下调的基因,颜色越深代表差异程度越明显(图1)。

图1 差异基因归一化处理后的热图
2.2 差异基因表达对获取的差异基因进行筛选,以基因表达值FC>1.5或<-1.5,标准化P<0.05作为筛选条件,得出筛选后的膀胱癌周围组织与原发膀胱癌差异表达基因共127个,其中在膀胱癌组织中上调的有4个,下调的有123个,对数据进行可视化处理,以火山图的方式呈现(图2)。
2.3 差异表达基因功能注释经GO富集分析和KEGG通路分析,我们发现膀胱癌肿瘤组织和癌旁组织样本的差异基因富集到的生物过程涉及细胞周期的调控、减数分裂的调控、肿瘤细胞的黏附机制和黏附分子合成、趋化作用、蛋白泛素化,其富集到的通路主要有Wnt/β信号通路、PI3K-AKt通路、MAPK通路、JAK-STAT通路等(图3)。
2.4 构建蛋白相互作用网络通过string蛋白互作数据库和cytoscape进行分析,筛选出相关核心基因,并从中挑选出最相关的6个基因进行分析。其中从癌旁组织相对上调的基因中筛选出MYL9、COL1A2、ACTG2,癌组织相对上调的基因中筛选出CDC20、ESM1、WDR72(图4)。
2.5 核心基因的验证通过GEPIA数据库进行生存分析,并且验证在不同分期膀胱癌组织中核心基因的表达情况。生存分析显示,MYL9、COL1A2、ACTG2的表达量与生存时间相关,差异具有统计学意义;在下调的基因中,CDC20、ESM1、WDR72其表达量与预后的关系不明确,差异无统计学意义(图5)。在膀胱癌和正常组织(图6)以及不同分期的膀胱癌组织中(图7),MYL9、COL1A2、ACTG2与肿瘤分期呈正相关,差异有统计学意义;ESM1表达量与肿瘤分期呈负相关,差异有统计学意义;CDC20与WDR72在不同分期的肿瘤组织中表达量的差异无统计学意义。

图3 核心基因富集通路的KEGG分析图
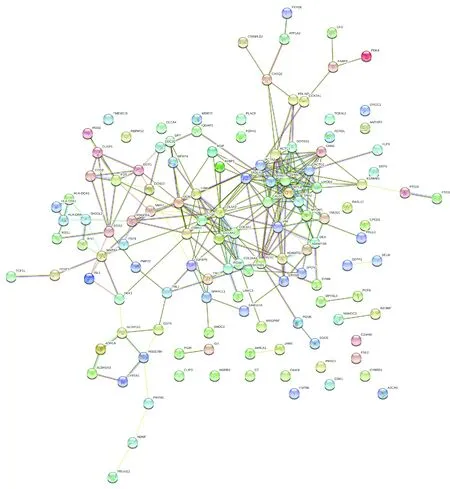
图4 蛋白互作网络图(圆形表示相关蛋白,直线表示相互作用关系)

A:COLA2;B:ACTA2;C:MYL9。
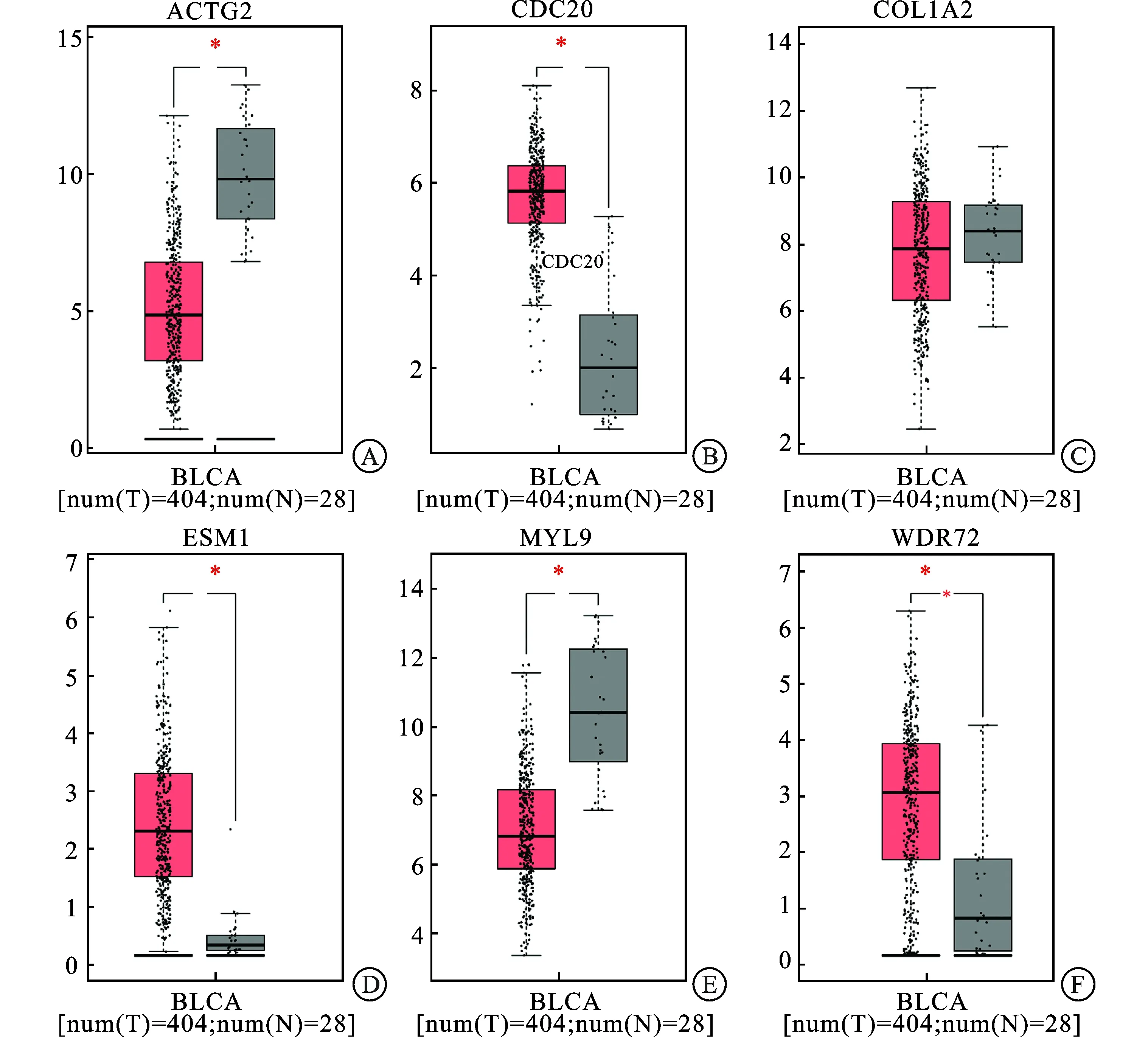
A:ACTG2;B:CDC20;C:COL1A2;D:ESM1;E:MYL9;F:WDR72。

A:ACTG2;B:CDC20;C:COL1A2;D:ESM1;E:MYL9;F:WDR72。
3 讨 论
在本研究中,我们对编号为GSE13507的基因芯片进行了差异分析,选用了FC>1.5或<-1.5作为纳入标准,并且选用P<0.05作为筛选标准,既避免了选用FC>2 或<-2,造成的潜在核心基因删失,又避免了制定过于宽松的纳入标准导致基因样本量过大。获得了膀胱癌组织与癌旁组织差异表达基因共127个,其中上调123个,下调4个。该芯片包含了较多的组织样本,从统计学的意义上较好的可信度。通过GO富集分析、KEGG通路分析与蛋白互作网络的构建,我们进一步缩小范围,得到了6个核心基因MYL9、COL1A2、ACTG2、CDC20、ESM1、WDR72,并且在GEPIA数据库中对他们进行了验证。再次挑选其中具有较好统计学意义的基因进行进一步分析讨论。
低表达基因MYL9编码肌球蛋白的轻链,同时还调控NMII的活性[1,7]。在细胞中它与肌动蛋白丝结合,能够控制细胞骨架构建,参与细胞形态的形成。同时它还通过调控粘附、迁移和信号转导因子,对肿瘤的侵袭和迁移产生重要影响[2]。有学者报道了通过组蛋白甲基化导致MYL9上调,从而在乳腺癌中介导了癌细胞的侵袭与转移,这对于研究MYL9在膀胱癌发生发展中的作用途径以及作用方式具有参考价值[8-9]。ACTG2编码的是肌动蛋白γ2,是一种高度保守的蛋白质,它与肌球蛋白一起参与各种类型的细胞运动并维持细胞骨架。目前已经在脊椎动物中发现了3种类型的肌动蛋白α、β和γ[5-9]。该基因在肠道疾病中的研究较为丰富。BUSS等[10]报道了ACTG2突变可导致先天性膀胱膨胀、微结肠和肠蠕动不全。该研究通过动物实验显示ACTG2存在于膀胱和肠道组织中转录本会干扰ACTG2的聚合,导致平滑肌收缩性受损。COL1A2基因编码Ⅰ型胶原蛋白的pro-alpha2链[11]。它由三重螺旋构成(2条α1链和1条α2链)。Ⅰ型胶原是存在于结缔组织中的纤维形成胶原,在骨、角膜、真皮和肌腱中含量丰富,与MYL9类似,对于肿瘤的黏附和侵袭有重要影响。
对于上调的基因ESM1,它可编码内皮细胞特异性分子内皮素,是一种表达于肺、肾内皮细胞等组织中的分泌蛋白[12-13]。该基因的表达受细胞因子调控,可能在内皮依赖性病变中起作用。肿瘤血管内的内皮素表达与肿瘤的分期和侵袭性密切相关,GEPIA数据库对不同的膀胱癌分期的ESM1表达分析的结果也与之相符。ROUDNICKY等[3]通过对ESM1转基因小鼠的机制研究发现,通过磷酸化VEGFR-2可以活化VEGF-A从而激活内皮素的表达,这一途径可以促进肿瘤血管生成,并导致预后不良。ESM1的数据库生存分析曲线也显示了这一点。目前ESM1在膀胱癌中的具体功能研究尚不明确,其更多机制有待进一步发现。
总之,我们利用生物信息学工具与基因芯片技术相结合,获取并筛选出差异基因,对其中的MYL9、COL1A2、ACTG2、CDC20、ESM1、WDR72进行了较为详细的生物学功能和分子机制分析。从基因层面研究了膀胱癌发生发展可能存在的机制,并且为进一步研究提供了可以供选择的治疗靶点和诊断靶点。
