神经发育疾病相关Tenascin C表达的分子调控通路
2021-04-12杨清湖何建秧白占涛
苏 琦,姜 鸣,4,杨清湖,4,杨 亮,4,何建秧,刘 霞,4*,白占涛,4*
(1.延安大学 生命科学学院;2.多肽资源药物研究中心;3.延安市特色资源生物工程技术研究中心;4.陕西省区域生物资源保育与利用工程技术研究中心,陕西 延安 716000)
胞外基质(Extracellular matrix,ECM)是细胞与细胞间的重要组成部分,由细胞合成、分泌到细胞外,介导细胞与细胞、细胞与细胞外的组织结构。神经系统ECM可形成具独特的神经元周围网络(Perineuronal nets,PNNs),包裹在神经元胞体和近端树突,参与神经元的形态结构、组织发育、分化迁移以及突触可塑性调控。PNNs主要成分为硫酸软骨素蛋白聚糖(Chondroitin sulfate proteoglycans,CSPGs)。根据硫酸软骨蛋白与氨基聚糖的不同,CSPG可分为Tenascin、Neurocan、Aggrecan、Versican、Phosphacan、Brevican等亚型[1]。
哺乳动物Tenascins(TNs)家族成员包括Tenascin C (TNC)、Tenascin R (TNR)、Tenascin W (TNW)、Tenascin X(TNX)、Tenascin Y (TNY)、Tenascin N (TNN)6种亚型,在脊椎动物高度保守。TNC是CSPG的主要组分之一,是由同源单链组成的六聚体细胞外基质糖蛋白,以核心架构分子组装PNNs。TNs的6条分枝整合胞外其它基质组分、细胞粘附因子,并与细胞膜锚定。
TNC表达呈现独特的时空特性。神经系统PNNs主要表达TNC和TNR,与神经发育与神经损伤修复、认知障碍、阿尔茨海默病(Alzheimer’sDisease,AD)神经退行性疾病密切相关[2]。从在发育过程中,TNC主要在胚胎发育中高度表达,主要分布于运动细胞周围、上皮间质相互作用和分支形态形成部位、致密结缔组织,如骨、软骨和肌腱;成年后,TNC仅高表达于肌腱、干细胞龛或发生应激损伤或病变组织细胞[3]。人源TNC表达量以阑尾中最高,其次为肾脏、肝脏、肺、脾脏、大脑等,而在啮齿类全脑胚胎发育及成年后的结肠、卵巢、膀胱高表达。但迄今,有关TNC时空差异表达与神经发育、神经疾病发生的相关机制,仍不完全清晰。本文聚焦TNC基因与转录调控元件、TNC蛋白组构的多样化功能结构域、TNC蛋白合成转运分泌的视角,解析神经发育疾病相关的TNC调控通路,以提出基于TNC作为组成型胞外基质或调节型功能表达的神经发育疾病诊疗新策略。
1 TNC基因与转录调控元件
人TNC基因位于9q33染色体上,包含29个外显子,转录起始由一个非编码外显子开始,这是由于在E1处存在一个转录起始位点TSS,相隔20 kb内含子后有一个包含翻译起始密码子的E2外显子。人和小鼠的TNC启动子都包含TATA盒,近端序列-39 bp~-1414 bp范围包含多个转录因子结合位点,其中TSS上游220 bp之内为高启动子活性区域,在此区域内有8类转录因子结合位点,在214 bp之后仅存在4个已知位点。目前在人类疾病范围被报道的有8类,包括ETS、SP1、SMAD、NOTCH、JUN、NFκB、GATA、OTX2,而以鼠源为背景报道的只有HBS、NF1、OCT、EGR1及-1414 bp的SRE/MKL1位点[4]。
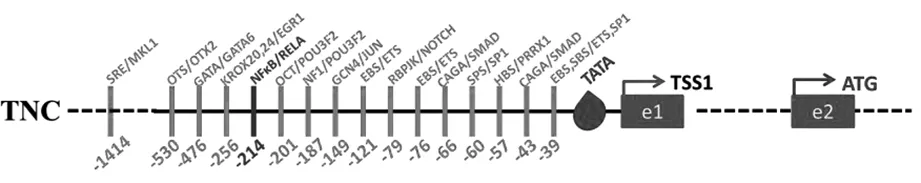
图1 TNC基因启动子元件组构
1.1 神经增殖与分化的ETS-TNC通路
ETS作为最大的进化保守转录因子家族成员之一,分别在TNC启动子近端区域的-39 bp/-76 bp/-121 bp处都存在结合位点。E26转化特异性(ETS)转录因子家族作为转录激活或抑制因子在胚胎发育和病理过程中参与细胞分化、增殖、迁移和凋亡,通过转录水平及蛋白磷酸化、糖基化调控其活性,以促进ETS靶基因表达。疾病发生后上游磷酸化信号通路异常是导致ETS家族激活的关键机制之一,从而调控TNC的表达[5]。
胚胎发育时期ETS促进神经元分化,轴突新建,ETS的有序转录可促进脊髓运动神经元(Motor neurons,MNs)的成熟分化及神经元的连通性,ETS提前启动可导致神经元分化异常,减少感觉神经元传入投射[6]。ETS家族成员Pet-1缺失可使5-羟色胺(5-Hydroxytryptamine,5-HT)合成、摄取和储存降低,导致成年后出现焦虑及攻击行为的概率升高[7];ETV5的去抑制作用有利于神经干细胞(Neural stem cells,NSCs)增殖和自我更新,减少NSCs向胶质谱系的选择,利于神经细胞的生成,从而控制少突胶质细胞瘤的发生[8]。ETS转录因子可直接与TNC启动子近端位点结合上调TNC的表达,并且ETS可关联SP1、JUN进一步增加自身与TNC的亲和力,转化生长因子(Transforming growth factor-β,TGF-β)可调控这一方式[9]。TGF-β首先与星形胶质细胞表面Ⅱ型受体及磷酸化Ⅰ型受体的GS域结合,胞内SMAD2/3发生磷酸化后与SMAD4结合转移至细胞核激活ETS2调节TNC的表达[10]。因此,以ETS因子为切口探究其靶基因TNC可为神经分化和细胞增殖提供依据。
1.2 神经损伤与疾病的JUN-TNC双调节方式
JUN转录因子家族成员c-JUN在发育及神经损伤中可控制神经细胞的死亡和退化、胶质增生、可塑性及修复[11]。c-JUN可通过c-JUN-N-末端激酶(JNK)信号通路及作为活化蛋白-1(AP-1)复合物成分两种方式发挥作用。c-JUN/JNK信号通路可促进细胞凋亡,调节神经系统发育、轴突再生和神经元变性,其中JNK1、JNK2对发育中的胚胎神经管凋亡和成年神经损伤后神经再生有促进作用[12]。神经损伤后的脊髓背角星形胶质细胞中c-JUN/JNK信号通路激活,JNK1磷酸化增多,并与TNC结合发挥作用[13]。
AP-1作为c-JUN的另一种主要作用方式参与诱导发育过程中BDNF外显子I、III和VI转录本,帮助调节神经元分化及突触可塑性的建立[14]。人类和小鼠组织中c-JUN和c-Fos组成AP-1复合物,参与细胞增殖、凋亡和组织形态形成,AP-1转录因子与DNA结合,调控靶基因TNC转录,并可接受细胞外信号传导引起转录活性和TNC表达的改变[15]。AD中JNK/AP-1通路的激活可促进Aβ样斑块周围的星形胶质细胞的吞噬,这可能与星形胶质细胞分泌的TNC有关[16]。c-JUN的高表达可激发TNC启动子活性,通过染色质免疫沉淀检测到AP-1富集于TNC启动子区域[17]。因此,JUN-TNC的双调节方式通过调节神经细胞程序性死亡及胶质细胞吞噬过程在神经损伤和疾病过程中发挥的生物学功能。
1.3 神经炎症的NFκB-TNC互作通路
NF-κB转录因子主要行使免疫系统应对炎症和感染的监管作用。胚胎发育中NF-κB敲除可诱发严重的发育缺陷,如神经元上皮细胞凋亡增加、神经管无法关闭,且NF-κB成分亚基p65缺失导致小鼠早期胚胎高的致死率[18]。小胶质细胞中TLR4/NF-κB信号通路及TNF-α/NF-κB通路激活可诱导TNC的瞬时高表达,引起神经炎性损伤[19]。在AD中TNC高表达反而可通过磷酸化Erk1/2使NF-κB激活,诱导其发生核易位,进一步激活凋亡相关的BCL家族,降低Caspase活性抑制凋亡,增强星形胶质细胞的清除吞噬功能[20]。因此,神经炎症发生中,NFκB-TNC存在功能性互作调控分子通路。
1.4 神经发育的TGF-β/SMAD-TNC通路
SMAD转录因子是TGF-β家族成员的关键效应因子,TGF-β/SMAD1/5/8信号通路可参与神经嵴细胞的发育、神经突生长,以及大脑皮层、海马、脊髓的多巴胺神经元发育、NSC向神经元和星形胶质细胞的分化[21]。AD中敲除TNC会使SMAD3减少,转录丰度降低,TGF-β/SMAD信号受损,降低神经元和胶质细胞SMAD2/3磷酸化及细胞核易位,通过调控TNC/SMAD信号通路可实现抑制小胶质细胞的激活,减弱神经毒性[22]。因此,TGF-β/SMAD-TNC通路可成为异常的神经发育及AD的重要诊疗策略。
1.5 神经发生的NOTCH-RBPJk-TNC的激活通路
NOTCH是一种跨膜蛋白,在胚胎和成人大脑的神经干细胞维护和神经发生中起关键作用,该信号激活后其胞内结构域裂解转移至细胞核,通过与转录辅助因子RBPJk结合发挥转录调节作用,其信号解除与神经退行性疾病及脑功能紊乱相关[23]。NOTCH受体1-4在不同类型的细胞中表达存在差异,神经元(Notch1和Notch2)、星形胶质细胞(Notch1和Notch2)、内皮细胞(Notch1和Notch4)及神经干细胞(Notch1和Notch2)中都有NOTCH受体的表达[24]。恶性胶质母细胞瘤组织中Notch2蛋白的高表达,以及RBPJk与TNC显著共表达指出NOTCH-RBPJk-TNC激活通路参与胶质细胞的异常增殖[25]。另外,该通路异常与成年海马的神经发生和认知障碍有关[26]。
1.6 胚胎发育的PRRX1-HBS-TNC互作通路
PRRX1在胚胎发育中期持续表达,定位于从多能祖细胞阶段到终末分化早期的整个生命过程中的细胞中,PRRX1缺陷可导致小鼠器官组织发育受损,在伤口愈合的细胞增殖过程中,关键因子Twist1的上调可与TNC启动子近端-57 bp位点的PRRX1结合增加TNC的表达,形成Twist1-PRRX1-TNC正反馈回路[27]。PRRX1还可直接与HBS因子相互作用激活TNC转录,参与细胞增殖[28]。胶质母细胞瘤中可通过PRRX1调节胚胎神经前体的自我更新和分化能力,并由多巴胺D2受体所介导[29]。
1.7 TNC表达的多转录因子协同调节
在调控TNC表达时还存在多种转录因子协同作用。成纤维细胞中,TGF-β除了诱导TNC表达以外,还参与SMAD3/4与启动子近端特异位点的结合过程,进而影响了SMAD3/SP1/ETS1形成转录复合物[30]。血小板衍生因子PDGF与成纤维细胞表面受体结合,调节TNC依赖的磷酸化PI3K-Akt通路,这一过程可由ETS1/2和SP1形成的复合体所介导[31]。另外,Sp1和SMAD转录因子协同介导TGF-β依赖性的Aβ前体蛋白基因转录、ETV5和RBPJK联合缺失可使小鼠胚胎干细胞(Mouse embryonic stem cells,ESCs)即使是在有分化刺激的情况下,依然保持未分化和自我更新状态[32]。与TNC启动子近端位点结合的NF-1、SP1、MLK1、POU3F2转录因子也参与神经发育或疾病过程。因此,探究TNC启动子近端与各个转录因子的结合以及多种因子的互作对神经发育疾病的治疗至关重要。
2 TNC蛋白的多样化功能结构域
2.1 TNC分子结构域多样性
随着各类胞外基质分子结构越来越清晰,TNs家族具有相同的分子结构,包括N末端富含半胱氨酸的TA(Tenascin assembly)结构域、表皮生长因子EGF(Epidermal growthfactor-like)样结构域、纤维连接蛋白FN(fibronectin type III-like)样结构域和C末端纤维蛋白原FG(fibrinogen-like)样结构域[33]。人类TNC是由2个三聚体形成的六聚体结构,每个TNC单体包含1个N末端的富含半胱氨酸的TA构域、14.5个EGF样结构域、8个FNIII样结构域和1个FG样羧基末端组成,其中FNIII5和FNIII6之间存在9个可变性剪接位点FNIIIA-D[34],凸显为TNC功能结构域的多样性。
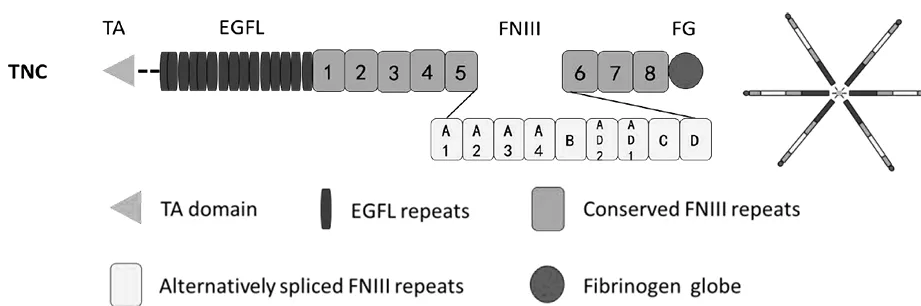
图2 TNC蛋白分子结构
2.2 TNC与膜受体、膜通道的结合域
TNC多样化功能结构域提供丰富的受体结合位点,血小板衍生生长因子(Platelet-derived growth factor,PDGF)家族、成纤维细胞生长因子(Fibroblastgrowthfactor,FGF)家族、TGF-β超家族和神经营养因子(Neurotrophin,NT)与TNC的FNIII1-5、FNIII12-14结构域,特别是FNIII5结构域具有高亲和力[35],EGFL重复域可作为表皮生长因子(Epidermal growth factor receptor,EGFR)的配体参与病理过程[36]。这种基于结构的作用方式可为探究TNC在神经发育和疾病的机制提供新理解。
由FNIII6重复序列编码的新型肽VSWRAPTA可通过激活胞外的局灶性粘附激酶(Focal adhesion kinase,FAK)和下游ERK1/2信号通路显著促进胚胎皮层神经元突起的生长及分支形成[37],α9β1整合素与FNIII3的结合还可影响间充质干细胞(Mesenchymal stem cells,MSCs)向神经元的分化[38]。另外,在调节突触可塑性时TNC可与整合素相互作用引起酪氨酸磷酸化、Ca通道开放,或者直接导致Ca通道开放调控可塑性,由TNC缺失引起的突触可塑性下降,可被L型钙通道拮抗剂硝苯地平逆转[39]。TNC的EGFL域与钠通道β亚基的胞外免疫球蛋白结构域的结合与神经元动作电位传导有关。这都提示TNC蛋白结构域多样性可通过膜受体和通道结合方式参与神经分化及突触可塑性调控。
2.3 中枢神经TNC合成、转运和分泌机制
TNC是在胞内合成转移至胞外成为ECM,分子结构域的多样化决定了TNC可通过配体-受体结合或跨膜通道方式转运至细胞内,胞内TNC的表达与其转录元件调控紧密相关。新生儿时期星形胶质细胞分泌大量TNC,随后自身EGFL域和NSC表面的EGFR结合转运至细胞内,激活PI3K-AKT通路促进NSC向神经元分化,实现了TNC在不同类型细胞间的转运[40]。星形胶质细胞内TNC的FNIII3结构域可与整合素α9β1、αVβ3、α8β1、αVβ6结合,下游靶点雷帕霉素(Mechanistic target of rapamycin kinase,mTOR)的激活后促进上调TNC表达,有助于大量TNC分泌至胞外参与神经炎症、损伤及神经退行性疾病[41]。研究已经发现AD中星形胶质细胞分泌的TNC可能参与周围Aβ斑块的解离,这一发现可为临床治疗AD提供新思路。但TNC是否是通过囊泡转运的方式实现向其他类型细胞的转运将是研究神经发育和神经退行性疾病的目标之一。
3 基于TNC调节型表达分子通路的神经发育疾病诊疗
无论是基于TNC转录元件调控还是分子结构域多样性的调控通路,都以调节TNC表达量为目的参与神经发育疾病。TNC的缺失往往导致胚胎期神经发育异常,并增加成年后神经疾病比率。胚胎发育期NSC增殖分化与TNC表达有关,TNC基因缺失可导致神经管无法关闭,并可通过减弱神经元连接及代谢诱发小鼠的认知障碍[41]。TNC在暴露于Aβ样斑块的小胶质细胞中基因转录增加而发生沉积,通过降低AD小鼠大脑中TNC的水平,可以有效降低小胶质细胞和巨噬细胞的数量,减少突触损伤[43]。TNC的敲除将神经炎症从原发性转变为抗炎性[44],通过降低AD小鼠大脑中TNC的表达,小胶质细胞和巨噬细胞的数量显著增加,Aβ蛋白与小胶质细胞及星形胶质细胞上的模式识别受体结合,触发以炎症介质释放为特征的先天免疫应答,从而加重疾病进程[45]。神经损伤后TNC表达上调可影响轴突再生[46],它作为一种内源性激活剂,可在促炎性细胞因子TGF-β、IL-6的作用下,在受损的AD大脑积累引起慢性炎症,突触可塑性降低。近期发现TNC敲除小鼠前额、海马的突触蛋白及星形胶质细胞标记物表达上调,这提示调节型TNC表达可通过影响神经元及胶质细胞参与神经发育疾病诊疗。
4 基于TNC胞外基质网络解离与重构的神经发育疾病诊疗
TNC作为中心构架参与PNNs形成,其中透明质酸形成PNNs的基本骨架,蛋白多糖连接蛋白(HAPLNs)稳定了透明质酸和硫酸软骨素蛋白多糖(CSPGs)之间的相互连接,CSPG核心蛋白的C端与TNC结合,从而形成具有独特网状结构的胞外基质网络,主要包裹大脑和脊髓PV+/GABA抑制性神经元的胞体和近端树突[47]。发育晚期PNNs开始形成,其发育成熟性和神经可塑性水平的高低呈负相关,PNNs解离往往导致癫痫、脑卒中、阿尔茨海默病、精神分裂症等神经功能障碍[48]。有研究指出敲除TNC会导致神经网络损伤、突触可塑性降低、认知行为改变,引起小鼠的空间学习障碍[49]。AD中铁离子大量积累引起自由基的增加,促进氧化应激的发生,PNNs的多阴离子特性,使其具有结合大量铁离子的能力,从而通过降低氧化应激来保护PV+神经元[50],而使用硫酸软骨素酶ABC(Chondroitinase ABC,ChABC)去除CSPG后Aβ蛋白表现出神经毒性;另一方面,PNNs包围在Aβ斑块表明,通过ChABC使其解离,可以达到降解作用[51]。这表明胞外基质网络的解离与重构可能对神经退行性疾病具有很高的治疗潜力。
