微囊藻毒素降解酶MlrA的结构功能分析
2021-04-09潘禹王华生詹鸿峰孙缓缓范超刘祖文闫海
潘禹,王华生,詹鸿峰,孙缓缓,范超,刘祖文,闫海
(1 江西理工大学土木与测绘工程学院,江西赣州341000; 2 北京科技大学化学与生物工程学院,北京100083)
引 言
随着水体富营养化现象加剧,淡水蓝藻水华暴发形势愈发严峻[1]。微囊藻毒素(microcystins, MCs)是水华暴发期间检出频率最高、危害最为严重的肝毒性蓝藻毒素[2-3]。MCs 由7 个氨基酸组成,结构通式 为 环 - (Ala(1)-X(2)-MeAsp(3)-Z(4)-Adda(5)-Glu(6)-Mdha(7)),见图1。由于非保守位置(X 和Z)可变L-氨基酸的不同,以及MeAsp基团和Adda基团的甲基化和去甲基化差异,MCs 可形成超过200 种同分异构体,以MC-LR和MC-RR毒性最大且含量最高[4]。
交替双键和环状结构使MCs 在自然环境中保持高度稳定,对多数化学药剂与常见蛋白水解酶具有抗性,甚至在煮沸或极端pH 条件下仍不变性失活[4]。尽管如此,已经有研究发现了部分水生细菌对MCs 具有生物降解活性。目前,已从生态系统中分离表征出70 多株MCs 降解菌[5],并通过DNA 文库筛选与基因异源表达鉴定出功能水解酶MlrABCD[6]。MlrA(或称为微囊藻毒素降解酶microcystinase)首先攻击MC-LR 的Arg-Adda 肽键,产生低毒线性产物(H-HN-Adda-Glu-Mdha-Ala-Leu-MeAsp-Arg-OH);接着胞内酶MlrB 识别并水解线性MC-LR 的Ala-Leu 肽键,生成四肽(Adda-Glu-Mdha-Ala-OH);最后胞内金属酶MlrC 将四肽的Adda-Glu 肽键水解,生成最终产物Adda。MlrD 暂推定为转运蛋白,负责将MCs 或降解中间产物跨膜运输至细菌细胞体内[7]。
作为MCs 细菌降解过程中关键蛋白酶,MlrA 在水生态系统的修复过程中具有潜在应用价值。Dziga 等[8]通过基因克隆在大肠杆菌中过表达MlrA,将其固定在藻酸盐珠粒中用作全细胞生物催化剂,可在短期内(3 d)稳定去除连续流湖水培养基质中的MC-LR。Dexter 等[9]在蓝藻光合自养宿主中异源表达MlrA,使其在半自然条件下催化稳定性大幅度提高,成功实现MCs 水污染的原位修复。然而,目前尚未阐明MlrA 的结构特征与催化机理,导致无法通过分子修饰或定向进化等技术使酶满足工业生产的实际要求(如催化活力、热稳定性、环境耐受性等)[10-12]。因此,本研究的主要目标为:(1)基于蛋白质同源关系,采用分子模拟构建MlrA 结构模型;(2)将MlrA 与底物进行对接,分析其结合模式与相互作用;(3)采用定点突变验证假定活性位点的必要性;(4)研究酶活性的影响因素,明确其催化机理。
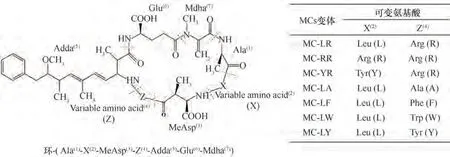
图1 MCs的结构通式Fig.1 The general structure of microcystins
1 实验材料和方法
1.1 材料
大肠杆菌BL21(DE3),质粒pET-30a(+),MCs 降解 菌Sphingopyxis sp. USTB-05 及mlrA 重 组 菌(GenBank:HM245411.1)均由本实验室保存[13]。
微囊藻毒素MC-RR(分子式C49H75N13O12, 分子量1038.2,95%)标准品购自台湾Algal Science 公司;乙腈(99.9%)、异丙基-β-D-硫代半乳糖苷(IPTG,99%)、酶抑制剂(EDTA、1, 7-菲咯啉和1, 10-菲咯啉,99%)购自Sigma Aldrich 公司。其余化学试剂均为分析纯。
1.2 模型构建
从NCBI 数据库检索Sphingopyxis sp. USTB-05菌的MlrA 序列(GenBank: ADK25053, FASTA 格式),使用ProtScale 工具分析氨基酸亲疏水性,采用PSIBLAST 在蛋白质结构数据库(PDB)中进行同源性分析。由于MlrA 与已解析结构蛋白的序列相似性较低(20%~30%),采用折叠识别法(fold recognition)进行分子模拟。首先通过Threading 程序(HHpred[13]、pGenTHREADER[14]、 Phyre2[15]、 SPARKS-X[16]和CEthreader[17])将MlrA 氨基酸序列提交至蛋白质折叠文库,筛选可用结构模板并生成序列-结构比对信息;随后提取所预测二级结构与模板蛋白进行比对;接着将对齐序列输入建模软件MODELLER 9.14,计算生成100 个原始模型并使用Loop refining模块对Loop 区域进行循环优化;最后选择具有最低DOPE得分和最高GA341得分的模型[18]。
基于TM-align 程序[19]计算的Cα-RMSD(均方根偏差)和TM-score 分布,从不同类模型中选择最佳模型,使用Deepview 在GROMOS96 43B1 力场中实施最速下降法进行能量最小化,使用PROCHECK[20]、ProSA-web[21]和ERRAT[22]分析蛋白骨架构型的几何质量、结构能量分布和残基相互作用。
1.3 分子对接
分子对接是通过计算模拟对两个或多个分子进行相互识别,使其满足几何匹配与能量匹配的过程。EDTA 三维分子结构取自NikA酶配体(PDB ID:1ZLQ);MC-LR 三维分子结构由ChemBioDraw 构建,使用UCSF Chimera 对其能量最小化,参数设置为:1000步的最速下降步骤和1000步的共轭梯度步骤,步长均为0.02 Å(1 Å=0.1 nm)。使用格点对接程序AutoDock 进行分子对接,其格点上为受体与探针原子间的相互作用能[23]。根据MlrA 活性口袋提取潜在催化位点(E172)的坐标,网格框参数设置如下:对接中心X = -105.85,Y = -8.85,Z = -88.05;盒子尺寸X = 60,Y = 60,Z = 60;格点之间的距离为0.375 Å。选择Lamarckian 遗传算法优化结合相互作用,共计算100 个结合构象。通过评估每种构象的结合自由能(ΔGbinding)与复合构象出现的频次及其合理性选择最佳结果在PyMOL和LigPlus中分析。
1.4 定点突变
全基因合成MlrA 突变体的DNA 序列并引入酶切位点,碱基替代结果见表1。使用Nde I和Not I酶切片段,将目的基因插入pET-30a(+)载体。将重组质粒转化至E. coli BL21(DE3)后涂覆于Luria-Bertani(LB)平板(含50 μg/ml 卡那霉素),挑取阳性克隆子并通过基因测序确认无误后进行下一步处理。
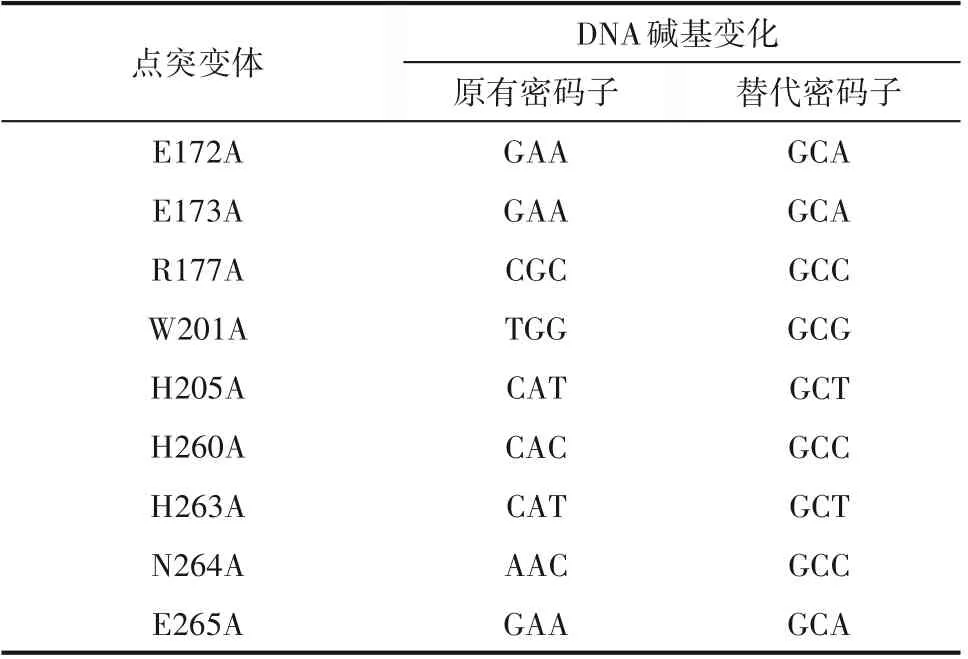
表1 MlrA点突变体的氨基酸残基及相应替代密码子Table 1 Amino acid residues and respective codons exchanged in MlrA mutant studies
1.5 MlrA的表达与提取
Sphingopyxis sp. USTB-05 与mlrA 重组菌按文献[24]方法培养。MlrA 突变菌培养方法如下:向含50 μg/ml 卡那霉素的LB 液体培养基中加入1%新鲜菌液,于37℃、200 r/min 转速下振荡培养至菌液OD600值达到0.6~0.8,加入终浓度为0.3 mmol/L 的IPTG 后降温至30℃诱导6 h。之后,将培养菌液离心(12000 r/min,20 min,4℃)收集,用PBS 缓冲液(pH=7.4)洗涤3 次。最后,将菌体用PBS 缓冲液重悬,于冰水浴中超声破菌,设定功率240 W,超声3 s间歇5 s,全程时间8 min,分3 次进行,以防止温度升高使酶变性失活。将破碎后的细胞再次离心,上清液含细菌全部可溶性蛋白。
1.6 in vitro酶活性分析
取定量酶液置于PBS缓冲液中,如有需要,分别与金属离子(Cu2+、Zn2+、Mn2+、Fe2+和Mg2+)或酶抑制剂孵育2 h,然后加入终浓度为10 mg/L MC-RR。制剂涡旋混匀后25℃反应60 min,加入10 μl 1% TFA 将酶灭活,经高速离心(12000 r/min, 10 min)后取上清液进行HPLC 分析。所有样品一式三份,计算样本的标准差(STDEV)反映结果相对于平均值的离散程度。
使用Agilent 1260型高效液相色谱仪和TC-C18反相色谱柱(4.6 mm×250 mm,5 μm)进行色谱分析。分析测定条件:流动相为含有0.05% (体积) TFA 的水(A)和乙腈(B)=65∶35(体积),流速1.0 ml/min;进样量20 μl;紫外检测波长239 nm;柱温箱温度35℃。
2 实验结果与讨论
2.1 MlrA分子模型构建
2.1.1 模板筛选 MlrA 由336 个残基组成,包含26个氨基酸编码的N 端信号肽[5-6,25],其序列下游为鉴别于CPBP 家族(CAAX proteases and bacteriocinprocessing enzymes,CAAX 蛋白酶和细菌素加工酶,其中C 是半胱氨酸,A 是脂肪族氨基酸,X 是任意氨基酸)的ABI(abortive infection)结构域[26]。结构模板初步筛选显示,PDB 数据库中缺少与MlrA 高度同源(≥30%) 的 蛋 白 质 晶 体 结 构,Methanococcus maripaludis 菌 Ras 转 化 酶 1(Ras and a-factor converting enzyme 1,Rce1)是唯一具有远程同源性的已解析蛋白(PDB ID: 4CAD)[27],序列同一性约为20%。Rce1 为定位于内质网的整合膜蛋白,介导CAAX蛋白羧基末端-AAX三肽的裂解,在细胞信号传导过程中具有重要作用[26-28]。由于具有多次跨膜结构,Rce1 直系同源物序列同一性(9%~63%)和分子大小(30×103~37×103)差异较大;但研究表明,任何类型同源物之间跨膜区段疏水性氨基酸存在差异是普遍现象[28]。因此,尽管Rce1 同源物相似性较低,但具有同样的催化结构。例如,MlrA和MmRce1都含有保守的ABI 结构域。考虑到两类蛋白同属CPBP 家族且具有膜相关性,MmRce1 为潜在可用结构模板。
通过比较MmRce1 与MlrA 的二级结构评估序列-结构比对的可靠性。根据图2 所示,MlrA 与模板结构高度一致,10 个α-螺旋与催化位点(红色标注氨基酸)的位置基本对齐。同时,比对信息还显示了部分差异:MlrA 在对齐结构中存在2 段缺口片段(Trp129-Ala136 和Thr271-Thr277),无 规 则 卷 曲的氨基酸形成了特殊环区(Loop 1,2)。上述差异将在模型分析部分中进行讨论。
2.1.2 模型建立与优化 通常,将序列同一性低于30%的目的蛋白称为过渡区(twilight zone)蛋白[29]。由于此类蛋白与模板蛋白之间的同源性较低,其结构预测是计算结构生物学的难点。Schoonman 等[30]研究表明,折叠识别法可在低同源性的情况下建立较为准确的结构模型。该方法不仅考虑了目的蛋白与模板之间的序列相似性,还以模板中的结构信息为基础,用以获取相似的折叠方式或结构基序[29]。基于此,本文使用5 种不同算法的Threading 程序分析了MlrA 与MmRce1 的最佳序列-结构比对,然后将各程序给出的比对结果提交至MODELLER,在空间约束下计算包含所有非氢原子的模型。最后,对模型的Loop 区进行循环优化,挑择满足最佳目标函数的模型并将其提交至TM-align 程序,根据TMscore 评估结构比对质量。初始模型的评估结果见表2。
根据表2 可知,由pGenTHREADER 构建的模型质量最佳,与原晶体结构的Cα-RMSD 值为0.68 Å,TM-score 为0.93,且对齐长度略长于其余模型。因此,选择该模型作为MlrA 最佳模型。为避免模型中存在不良分子接触,在GROMOS96 43B1 力场中实施最速下降法进行能量最小化。使用多个模型评估服务器验证优化模型的质量,结果见图3。PROCHECK 用于测量模型中逐个残基的立体化学质量。由图3(a)的Ramachandran 图发现,MlrA 中92.1%的残基位于有利区域(黑色),7.5%的残基位于允许区域(深灰色和浅灰色),仅有Val278 位于异常区域(白色)。由于该残基远离蛋白结合位点,其影响可忽略不计。使用ProSA-web 计算MlrA 模型结构的整体质量,其Z评分为-3.19,见图3(b)。对于相似大小的天然蛋白质,该分值表明模型质量在可靠范围内。使用ERRAT 程序检验了模型中非键原子相互作用的统计信息。如图3(c)所示,MlrA 模型的ERRAT 得分为89.30,表明骨架构象和非键相互作用合理(>50)。
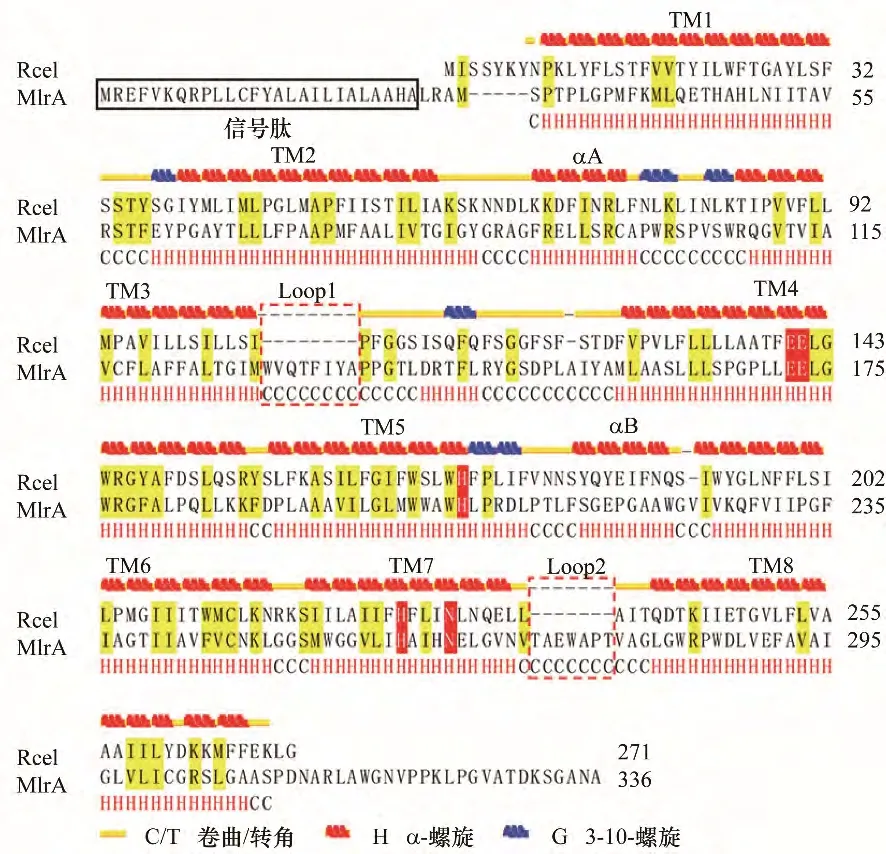
图2 MlrA和MmRce1的二级结构比对Fig.2 The alignment between MlrA and MmRce1 in secondary structure and residue levels
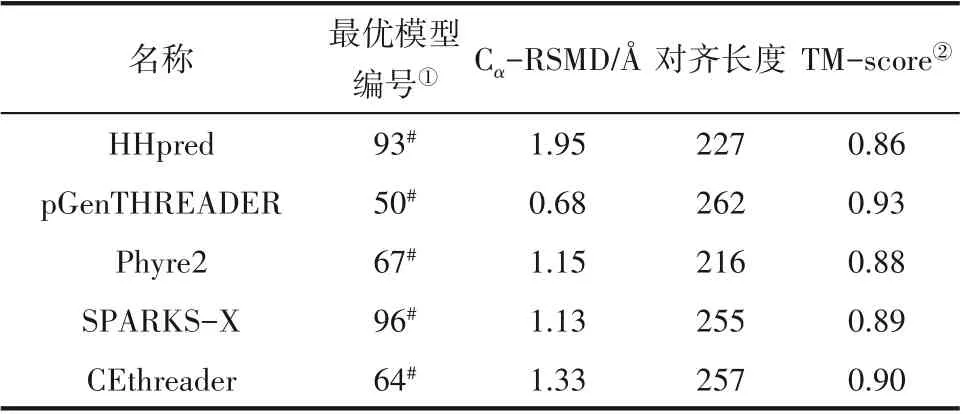
表2 不同Threading程序对齐结果模型的评估分析Table 2 Evaluation analysis of the models form the alignment results from the different Threading programs
2.1.3 模型分析 MlrA 主要由8 个保守跨膜螺旋(TM1~8)组成,螺旋之间由Loop 和膜外螺旋(αA 和αB)连接,见图4。MlrA 定位于细菌细胞质膜,使用MEMSAT-SVM 预测其N 末端和C 末端指向细胞质。与MmRce1 相比,MlrA 分子表面两个不同位置由较长的Loop 组成,在图4(a)中用黑色箭头指出。表面环区Loop 1 (Trp129-Ala136)增加了二级结构之间的运动幅度,可能影响酶的稳定性[31]。而Loop 2(Thr271-Thr277)位于活性位点的U 形入口处,可能通过相互作用形成活性结构有助于底物催化[32]。约100 个残基组成的ABI 域对应于TM4~7,见图4(b)。4 个TM 螺旋交错形成反平行螺旋束,被TM1~3 和TM8 螺旋包围,使内部形成体积较大的锥形催化空腔,且腔体一侧通过TM2 和TM4 之间的间隙向外敞开。上述构型允许溶剂与底物从空腔底部的周质空间进入酶分子内部,与活性位点进行相互作用[27]。
MlrA 的拓扑结构与氨基酸亲疏水性见图4(c)。与真核生物类似,细菌所表达的膜蛋白同样依靠信号肽系统进行定位跨膜。MlrA 在细菌胞质内合成时,其N 端疏水且带正电的信号肽形成α-螺旋插入磷脂双分子层中;随后,具有反平行结构的跨膜螺旋TM1 固定在质膜上,使后续肽链准确跨膜。另一方面,通过亲水性指数(数值越低则亲水性越强)可判断出MlrA 的跨膜区段具有强疏水性,这可能是其在各类载体(pGEX-4T1[9,13],pGEX-5T[9],pET-21a[25],Synechocystis PCC 6803[9])中表达为不溶性包涵体的主要原因。此外,疏水性多肽会抑制核糖体翻译过程,甚至破坏宿主菌的原有细胞膜结构影响菌体生长过程。如Dexter等[9]和Dziga等[25]发现MlrA异源表达构建体的细胞生长速率大幅降低。
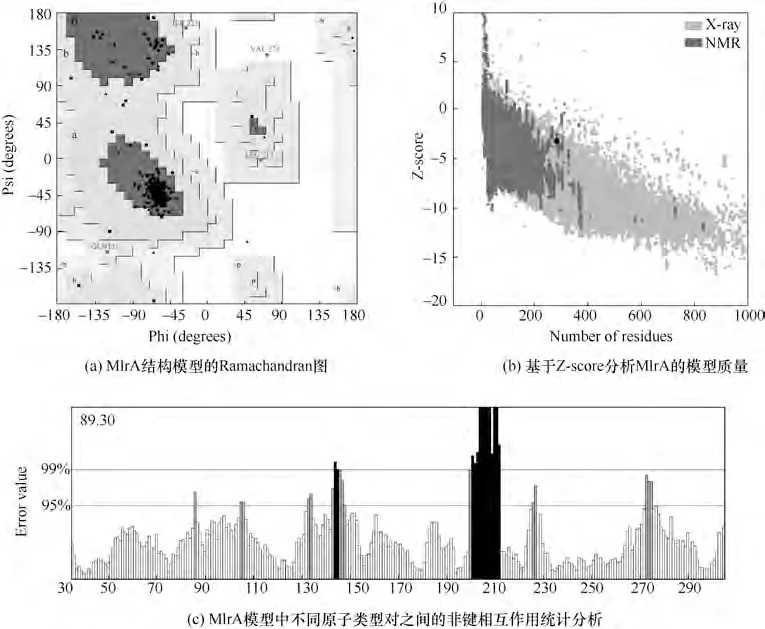
图3 MlrA分子模型质量检验结果Fig.3 Validation results for the MlrA model
通过结构叠加分析了催化位点的保守性。由图4(d)可知,MlrA 假定催化残基及水分子(W)的位置与MmRce1 活性位点呈良好重叠。图4(e)显示了活性位点的位置信息。E172、E173、H205、H260 和N264 均将其侧链投射至腔体内部,且原子间距在2.9~3.5 nm 之间,可形成氢键直接参与底物的催化反应。R177 位于催化腔顶部,可与E173 相互作用以阻止底物进入细胞质;而W176和W201的疏水基团埋藏在分子内部,表明上述残基不直接参与底物催化[27]。
2.2 MlrA底物水解机制
为了解MlrA 底物识别机制,以该酶的锥形催化空腔为活性口袋,研究了与MC-LR 的对接模式。MC-LR 单体的球棍模型如图5(a)所示,根据对接过程中所获的结合分数(ΔGbinding= -5.42 kcal/mol,1cal=4.18 J)与合理性选择最佳构象,见图5(b)。MC-LR通过化学键扭转采用β-发夹构型(β-hairpin structure)进入MlrA 催化腔,以降低底物基态与过渡态之间的能差。除Adda 侧链外,MC-LR 剩余基团将非极性氨基酸修饰的TM2 和TM4 之间的空间填充,从而使可断裂键Adda-Arg 暴露于活化水分子(W)与E172和H205附近。
图5(c)为MlrA 与MC-LR 之间的相互作用。位于相反TM螺旋的E172和H205彼此相对,桥连水分子构成谷氨酸(酸)—组氨酸(碱)—水分子(亲核)催化三联体。酸—碱—亲核三联体是共价催化过程中的常见基序,催化残基通过氢键作用建立电荷中继网络,以极化并激活亲核试剂攻击底物形成共价中间体,然后将其水解以释放产物并再生游离酶[27-28,33]。据此,提出MlrA 对MC-LR 的水解机理如图5(d)。即E172(酸)对齐并极化H205(碱),通过碱催化作用将水分子(亲核试剂)脱去质子激活,降低pKa对Adda-Arg 肽键进行亲核攻击并产生含氧阴离子中间体。H260 和N264 位于TM7 的连续螺旋转角,与催化二元组E172 和H205 相对应,通过氢键形成氧阴离子穴(oxyanion hole)使电荷在过渡态氧阴离子上聚集,以稳定产生的四面体中间体。中间体形成时Adda-Arg 肽键中的氮变为碱性,将H205 或E172质子转移至胺离去基团,迫使氧阴离子发生崩解产生酰基-酶中间体,使MC-LR 裂解为线性。此外,两个保守的芳香族残基(W176 和W201)与E172的侧链相接触,可能会提高谷氨酸的pKa,加速底物水解速率。
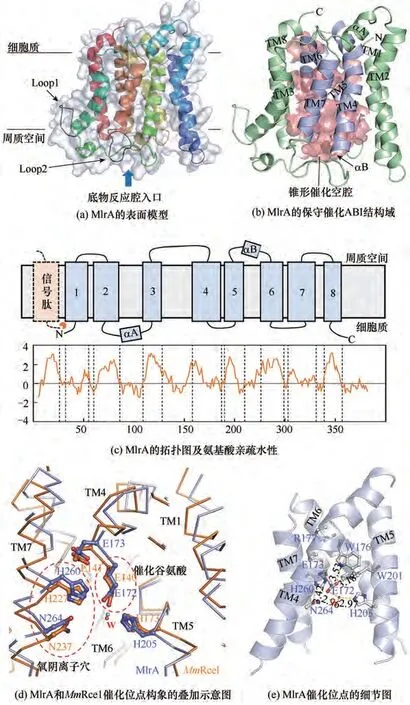
图4 MlrA分子模型的整体结构Fig.4 The overall structure of the MlrA model
2.3 定点突变
通过质粒定点突变,将假定催化残基替换为丙氨酸(A)验证了其催化必要性。由图6 可知,MlrA 活性依赖于E172、H205、H260、N264 和E265,其突变体导致酶活性完全丧失。催化三联体E172、H205和H260 的 重 要 性 已 在Rce1p[34],yRce1p[35]和MmRce1[27]中得到证实。类似于MmRce1,用Ala 取代N264 同样消除了酶活性,这表明N264 的去质子化作用可以使MlrA 失活,其羟基基团在催化过程中具有重要作用。另一方面,尽管E265 并未识别为MlrA 的活性位点,但其突变体对MC-RR 亦无降解活性,类似发现已在Dziga 等[25]研究中描述。由于谷氨酸具有负电性,推测E265可能作为质子供体将质子转移到反应的过渡态中间物,从而进一步稳定过渡态氧阴离子,使反应活化能降低并促进底物催化。
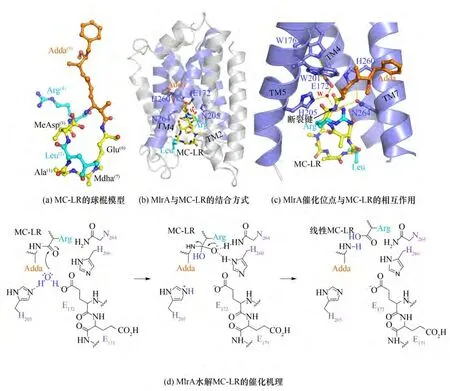
图5 MlrA与MC-LR的识别过程与催化机理(虚线表示氢键)Fig.5 Mode of action and catalytic mechanism of MlrA for MC-LR(dashed bonds represent the hydrogen bond)
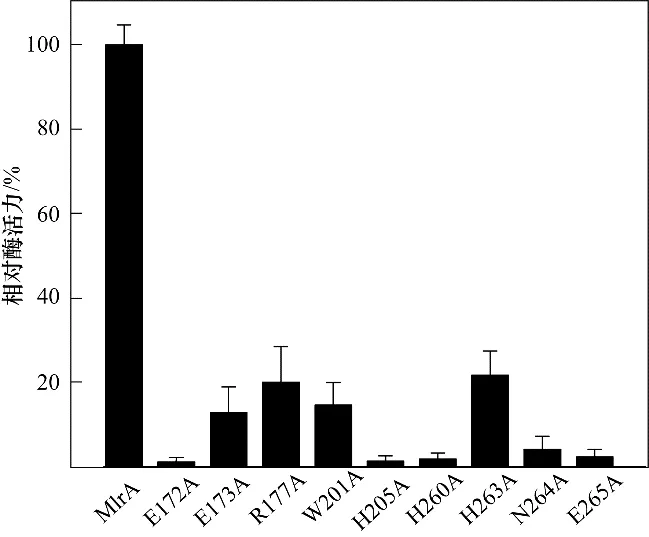
图6 MlrA与点突变体对MC-RR的催化活性Fig.6 Biodegradation activity of MlrA and point mutants for MC-RR
此外,E173A,R177A,H263A 或W201A 突变体会使MlrA 活性显著下降,这些残基位于功能基序附近,其侧链基团可能通过复杂相互作用与催化残基相接触,从而影响催化反应pKa。
2.4 MlrA活性影响机制
研究发现[7,25],金属螯合剂1, 10-菲咯啉和EDTA 可抑制MlrA 活性,且Bourne 等[6]提出其序列中含有假定的变体锌结合基序“HAIHNE(HEXXH)”。因此,MlrA 被推定为金属蛋白酶。然而,本研究通过分子结构模拟发现该酶不与Zn2+结合,因而底物水解机制不涉及金属基。此外,将MlrA 与金属离子共同孵育后评估了其剩余相对酶活,结果见图7,过量Zn2+和Cu2+使酶活性被完全抑制,Fe2+、Mg2+和Ca2+也不同程度降低催化效果,这初步表明金属离子(Ⅱ)无法与MlrA形成有效配位键使其活性提高。
为分析金属酶抑制剂对MlrA 失活原因,使用不螯合Zn2+的1, 7-菲咯啉(1, 10-菲咯啉的立体异构体)进行了酶活性分析。由图7 可知,两类化合物对酶的失活效果相似,浓度愈高抑制作用愈强,且在1 mmol/L 1,10-菲咯啉孵育后加入适量Zn2+无法恢复其催化活性(数据未显示)。显然,MlrA 活性与菲咯啉浓度相关,而不是Zn2+依赖性结果。Manolaridis等[27]和Dolence 等[35]研究表明,高浓度菲咯啉将引起MmRce1 和yRce1p 发生非特异性蛋白质解折叠现象,导致酶活力降低或丧失。由于MlrA 与Rce1 蛋白具有同源结构,其菲咯啉浓度依赖性失活机制相似。
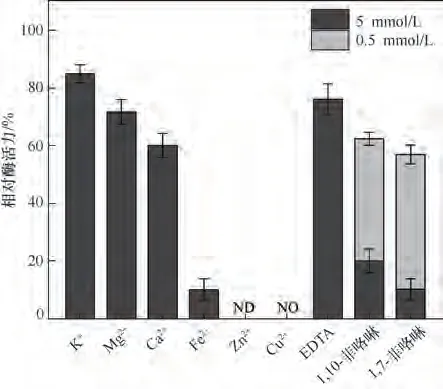
图7 金属离子和螯合因子对MlrA活性的影响Fig.7 Effects of metal ions and chelating factors on the activity of MlrA
另一方面,尽管MlrA 活性受EDTA 抑制,但作用机制可能不是结合酶分子中的金属离子。例如EDTA 通过竞争底物结合位点使人肝精氨酸酶[36]和TaqDNA 聚合酶[37]等非金属酶活性降低。图8 为EDTA 与MlrA 的结合方式(ΔGbinding= -3.47 kcal/mol)与相互作用,EDTA 结合在HAIHNE 基序附近,并与MC-LR 具有相似作用方式。LigPlot 分析表明,EDTA 羧基侧链与酶活性位点H205、N264 和S215、H263、R284 具有氢键结合作用;底物结合位点W201与G234、F235、G238与EDTA有疏水接触。综上,EDTA 主要通过竞争MlrA 活性位点降低其对MCs的催化效率。
3 结 论
(1) MlrA 是定位于细菌细胞质膜的整合膜蛋白,其分子结构中不含结合Zn2+,由ABI 结构域(TM4~7)通过反平行螺旋束形成向周质空间开放的催化空腔,使溶剂与底物可进入活性位点。
(2) MlrA 的 关 键 催 化 残 基 包 括E172、H205、H260 和N264。对MC-LR 水解机制主要为E172 和H205 桥接水分子形成酸(谷氨酸)—碱(组氨酸)—亲核(水分子)催化三联体,通过碱催化作用将水分子去质子化激活,以降低亲核残基的pKa对Adda-Arg肽键进行攻击。其次,H260 和N264 形成氧阴离子穴,提供氢键稳定所产生的四面体氧阴离子中间体。最后,H205 或E172 催化Adda-Arg 肽键中的胺离去基团发生质子化,使氧阴离子中间体崩解并将MC-LR裂解。
(3)MlrA 为非金属酶,不与金属离子(Ⅱ)配位结合,菲咯啉类化合物将导致酶分子发生非特异性解折叠而失活,EDTA 通过竞争酶活性位点降低催化效率。
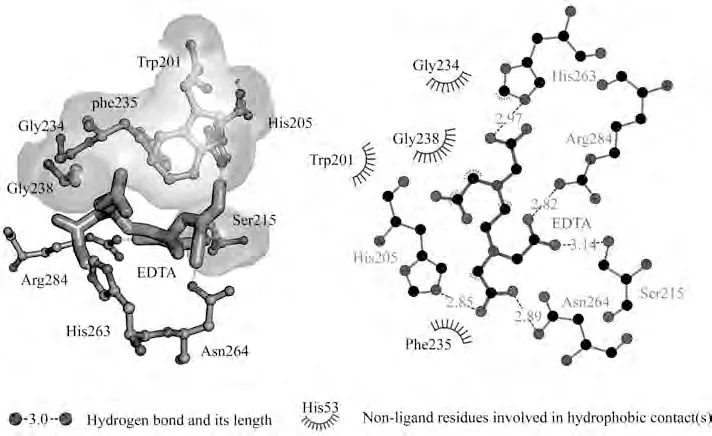
图8 MlrA与EDTA的相互作用Fig.8 The interaction networks between MlrA-EDTA complexes obtained by docking
