硫氮化合物在磷改性NiW/Al2O3加氢催化剂上的吸附行为研究
2021-04-09魏强黄文斌周亚松
魏强,黄文斌,周亚松
(中国石油大学(北京)重质油国家重点实验室,北京102249)
引 言
焦化蜡油等劣质重油中硫氮化合物含量高,结构复杂而且种类繁多,如果直接将其作为加氢裂化或催化裂化等工艺的原料,会导致裂化催化剂中毒以及裂化产品质量差等一系列问题,因此,劣质重油的加氢处理成为了石油化工领域最关键的技术之一[1-3]。硫化物中硫原子的脱除包括加氢脱硫路径(HYD 路径)和直接脱硫路径(DDS 路径)两种方式,前者为硫杂环和芳环加氢饱和后再进行C—S键氢解断裂,后者则是C—S 键直接断裂脱除硫原子[4-5]。对于氮化物而言,不管是吡啶类的碱性氮化物还是吡咯类的非碱性氮化物,其氮原子的脱除都以加氢脱氮(HDN)路径进行,即先发生氮杂环和芳环的加氢饱和,再进行C—N 键的氢解断裂[6]。不论以何种方式脱除杂原子,硫氮化合物都需要先克服扩散阻力进入到加氢催化剂孔道内,再吸附于载体或活性相上并发生加氢和氢解反应[7—8]。因此,研究硫氮化合物在加氢催化剂上的吸附行为是探究杂原子脱除机理以及加氢催化剂设计与制备的基础。
加氢催化剂一般由载体和负载在载体表面的活性相共同构成,载体主要为Al2O3,活性相一般由ⅥB 族的主金属Mo、W 以及Ⅷ族的助金属Co、Ni 的硫化物[Ni(Co)W(Mo)S]共同构成,二者相互协同以发挥较高的活性[1,9-11]。然而常规Al2O3表面酸性单一,只有L酸,且酸性较强,并且与活性金属之间的相互作用过强而不利于活性金属的分散和堆垛[6,12]。众多学者基于此以磷为改性剂研究了磷对Al2O3加氢催化剂理化性质及加氢性能的影响[13-16]。Tong 等[14]研究发现,磷的添加可以增强Ni 和W 活性金属在Al2O3载体表面的分散,并增大了催化剂平均孔径以及弱酸中心数量,当磷添加量为1.0%~1.5%时催化剂生成了更多的Ni-W-O 相,从而提高了催化剂的加氢活性。Tao 等[15]研究了喹啉和吲哚对噻吩类含硫化合物在NiW/Al2O3催化剂上加氢脱硫(HDS)的抑制作用,结果发现氮化物的种类和含量是影响硫化物HDS 的重要因素,在不同的实验条件下硫氮化合物分别表现出不同的竞争关系。王倩等[16]以喹啉和吲哚为模型化合物,考察了二者对二苯并噻吩(DBT)在NiW/Al2O3催化剂上HDS 活性和选择性的影响。研究表明,当氮化物存在时,DBT 的DDS 路径选择性得到提高,而HYD 路径受到较大程度抑制。因此,虽然目前已有大量关于硫氮化合物在磷改性Al2O3加氢催化剂上的反应研究,但大都未能较好阐述磷的引入对硫氮化合物吸附行为的影响。
本文以碱性氮化物喹啉、非碱性氮化物吲哚以及DBT 为模型化合物,通过考察其单独或同时在Al2O3及P/Al2O3系列催化剂上的吸附行为,探究了喹啉、吲哚和DBT 吸附行为与催化剂理化性质以及吸附质本身性质的关系,为硫氮化合物的吸附机理以及加氢催化剂的设计与制备提供参考。
1 实验材料和方法
1.1 Al2O3载体及P/Al2O3载体的制备
Al2O3载体的制备:取一定量的拟薄水铝石为铝源,去离子水与拟薄水铝石粉末质量比为:1.3~1.4,浓硝酸:4%,田菁粉:2.5%,柠檬酸:5%(均为质量分数)。其中田菁粉和柠檬酸作为复合助挤剂,田菁粉可以提高挤条速度,柠檬酸可以调高挤条的强度;浓硝酸作为胶溶剂,可以提高载体强度,改善孔结构。首先将挤条溶液(去离子水、浓硝酸和柠檬酸)与挤条粉(拟薄水铝石和田菁粉)混合,然后在F-26 双螺杆挤条机上混捏3 次使其混合充分,最后加孔板进行挤条成型。将挤条样品晾干后,再于120℃下干燥12 h、550℃下焙烧6 h制得成型Al2O3载体。最后将成型Al2O3载体进行研磨和筛分,取0.250~0.425 mm的颗粒作为催化剂载体材料。
P/Al2O3载体的制备:以(NH4)2HPO4为磷源,按负载量计算并称取一定质量的(NH4)2HPO4溶于适量去离子水中,采用等体积浸渍法,将溶液滴加到0.250~0.425 mm 的Al2O3载体上。然后室温静置24 h,120℃下干燥4 h,干燥后的试验样品置于马弗炉中于550℃焙烧4 h。
1.2 催化剂的制备
本论文采用等体积浸渍法将活性金属负载于Al2O3载体和P/Al2O3载体上,制备成两个系列催化剂,即:Al2O3系列催化剂和P/Al2O3系列催化剂。活性金属负载量(质量分数)为NiO:4%,WO3:26%。具体制备方法如下。
(1)称取一定量的Al2O3载体和P/Al2O3载体,将适量的硝酸镍和偏钨酸铵固体溶于适量的去离子水中配制成浸渍液,采用等体积共浸渍法将浸渍液滴加到相应的载体上。
(2)将样品在室温下充分晾干后,放入120℃烘箱中干燥4 h,最后将其放入500℃马弗炉中焙烧4 h,制得的两个系列催化剂。Al2O3系列催化剂:Al2O3、Ni/Al2O3、W/Al2O3、NiW/Al2O3;P/Al2O3系 列 催化剂:P/Al2O3、Ni/P/Al2O3、W/P/Al2O3、NiW/P/Al2O3。
1.3 催化剂表征
N2物理吸附-脱附表征:催化剂样品的孔道结构特征(比表面积、孔体积以及孔径分布等)参数在美国Micromeritics ASAP 2010 物理吸附仪上测定。将催化剂样品在300℃真空条件下脱气处理4 h,然后冷却至-196℃进行N2吸脱附,得到N2吸脱附量随体系压力的变化情况(体系压力变化范围为0~0.1 MPa),最后以吸附支曲线为准采用BET方程计算催化剂样品比表面积,以BJH 法计算催化剂样品的孔体积和孔径分布。
X 射线粉末衍射(XRD)表征:催化剂上的金属氧化物的分散度及晶型测定采用Rigaku D/MAX3400 型多晶粉末衍射仪,光源波长1.54 nm,扫描范围2θ为5~80°,扫描速度为2°·min-1。
NH3-TPD 表征:催化剂表面酸性测定在实验室自组装仪器上进行。称取300 mg 样品于石英管内,用高纯氮气作为载气,将样品在氮气氛中以10℃·min-1的速率从室温升至120℃,恒温30 min 后切换成NH3吸附至饱和。然后在氮气氛中以10℃·min-1的升温速率从100℃升温至500℃,通过TCD 检测脱附信号,用实验室SP-1000色谱记录酸量变化曲线。
吡啶吸附红外光谱(Py-IR)表征:催化剂表面酸类型及含量通过Magna-IR 560 E.S.P 型红外光谱分析仪测定。仪器的分辨率为4 cm-1,扫描次数80次。所测样品经真空升温处理后,降至室温,吸附饱和吡啶蒸气30 min,随后分别程序升温到200℃和350℃,并在此温度下脱附0.5 h,脱附完成后令其自然冷却至室温,扫描谱图,得到样品的吡啶红外光谱图。根据Py-IR 谱图中红外峰的位置和积分面积可计算各催化剂样品表面不同强度的B 酸和L 酸酸量。
1.4 吸附实验测定方法
1.4.1 动态吸附实验测定法 将喹啉、吲哚、二苯并噻吩(DBT)的环己烷溶液作为吸附实验的模型化合物。环己烷溶液中的氮或硫含量均为1000 mg·L-1。将3.895 g 催化剂样品(Al2O3系列催化剂和P/Al2O3系列催化剂)置于100 ml 容量瓶中,加入50 ml配制好的含有氮化物或硫化物的环己烷溶液,置于40℃的恒温水浴中并定时震荡。每隔一定时间取微量试样于色谱瓶中,然后用化学发光定氮仪或荧光定硫仪分析溶液中氮化物或硫化物的浓度变化,吸附量通过式(1)计算得出[17]:
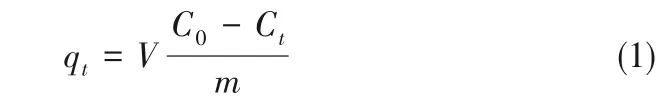
式中,qt为t时间吸附剂对吸附质的吸附量,mg·g-1;V 为溶液体积,L;C0为溶液初始浓度,mg·L-1;Ct为t时间溶液的浓度,mg·L-1;m为吸附剂的质量,g。
1.4.2 静态吸附实验测定法 将喹啉、吲哚、二苯并噻吩(DBT)的环己烷溶液作为吸附实验的模型化合物。环己烷溶液中的氮或硫含量均为1000 mg·L-1。将3.895 g 催化剂样品(Al2O3系列催化剂和P/Al2O3系列催化剂)置于100 ml 容量瓶中,加入50 ml配制好的含有氮化物或硫化物的环己烷溶液,置于40℃的恒温水浴中,静置48 h 后取微量试样于色谱瓶中,测定溶液中氮化物或硫化物的浓度,平衡吸附量通过式(2)计算得出[17]:
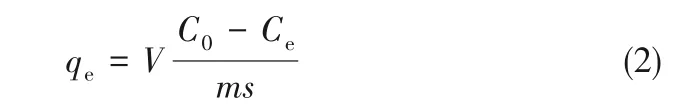
式中,qe为平衡吸附量,mg·m-2;V 为溶液体积,L;C0为溶液初始浓度,mg·L-1;Ce为平衡浓度,mg·L-1;m 为吸附剂的质量,g;s 为吸附剂的比表面积,m2·g-1。
2 实验结果与讨论
2.1 Al2O3及P/Al2O3系列催化剂的孔结构表征
Al2O3及P/Al2O3系列催化剂的孔径分布曲线如图1 所示。从图中可以看出,Al2O3系列催化剂的孔径分布集中在4~9 nm之间,而P/Al2O3系列催化剂的孔径分布集中在5~10 nm 之间,说明经磷改性后Al2O3系列催化剂的孔径有所增加,这是因为磷的引入堵塞了Al2O3部分微孔,增大了较大孔径的比例,从而使得孔径分布曲线呈现向大孔径方向移动的趋势[18]。
Al2O3及P/Al2O3系列催化剂的孔结构性质如表1所示。由表1 数据可以看出,Al2O3系列催化剂经磷改性后其比表面积和孔体积都有所下降,而平均孔径有所增大,这也是因为磷的引入堵塞了氧化铝部分微孔,改变了其孔道结构,而当比表面积较小的P/Al2O3负载活性金属后,其微孔更容易被堵塞,因此使得平均孔径增加。Al2O3及P/Al2O3载体分别负载4% NiO、26% WO3以及同时负载4% NiO 和26%WO3后,其比表面积、孔体积都有很大程度上的降低,这主要是因为活性组分浸渍时形成的活性组分颗粒,将氧化铝部分窄孔道阻塞,导致随着金属总量的增加从而使得催化剂的比表面积、孔体积大幅下降[19]。
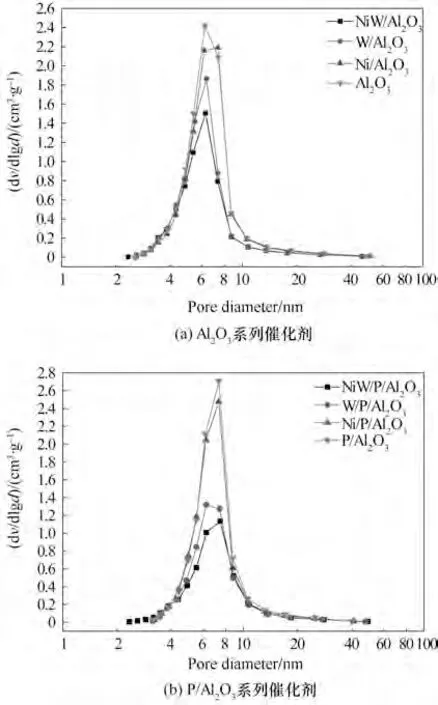
图1 Al2O3及P/Al2O3系列催化剂孔径分布曲线Fig.1 Pore size distribution of Al2O3 catalysts and P/Al2O3 catalysts

表1 Al2O3及P/Al2O3系列催化剂孔结构性质Table 1 Pore structure of Al2O3 catalysts and P/Al2O3 catalysts
2.2 Al2O3及P/Al2O3系列催化剂的XRD表征

图2 Al2O3及P/Al2O3系列催化剂的XRD谱图Fig.2 XRD patterns for Al2O3 catalysts and P/Al2O3 catalysts
图2为Al2O3及P/Al2O3系列催化剂的XRD谱图,谱图中γ-Al2O3(PDF No.29-1486)的特征峰位于2θ=37.2°、45.9°和66.8°处,而WO3体相晶体的特征峰位于2θ=23.7°、29°和33.6°处。根据图2(a)可以看出,Ni/Al2O3与Al2O3谱图并没有显著差异,说明NiO 在氧化铝载体上分散性较好,并没有出现较强的特征峰。从W/Al2O3谱图可看出WO3晶体特征峰,这说明此时催化剂中的WO3晶体颗粒尺寸较大,WO3分散性较差。当NiO 与WO3共同浸渍时,WO3分散度并没有发生明显变化。在图2(b)中,Ni/P/Al2O3与P/Al2O3谱图也没有显著差异,说明NiO 在P/Al2O3载体上分散性较好,并没有出现较强的特征峰。结合图2(a)和(b)不难发现,磷的引入使W/P/Al2O3表面羟基相互作用,消除了WO3与载体强相互作用的作用位,导致在焙烧过程中WO3易发生团聚形成体相晶体,所以WO3体相晶体的特征峰突出明显。而当P/Al2O3载体同时负载NiO 和WO3时,WO3体相晶体的特征峰又消失了,这可能是因为NiO 的引入提高了WO3的分散度,同时降低了WO3晶体尺寸[20-21]。
2.3 Al2O3及P/Al2O3系列催化剂的NH3-TPD表征
根据NH3脱附的温度,可将酸中心类型分为三类:150~250℃脱附时为弱酸中心;250~400℃脱附时为强酸中心;400~500℃脱附时为强酸中心。Al2O3及P/Al2O3系列催化剂的NH3-TPD 谱图如图3 所示。从图3(a)中可以看出,只负载NiO 的Ni/Al2O3催化剂表面酸量与Al2O3载体相比变化不大,说明NiO 在载体表面并没有提供较多的酸中心数量。而负载WO3的催化剂表面酸量与载体相比,提升幅度较大,说明活性金属WO3在载体表面提供了较多的酸中心数量。对于同时负载NiO 和WO3的催化剂而言,其酸中心数量介于Ni/Al2O3催化剂与W/Al2O3催化剂之间而远远超过Al2O3载体。对比图3(a)、(b)不难发现,磷的引入能够有效提高Al2O3系列催化剂表面的弱酸中心数量,这是因为磷与Al2O3载体表面铝羟基作用形成了更多酸性较弱的磷羟基。而对于W/Al2O3催化剂而言,磷降低了其强酸中心数量,这也是因为磷消除了WO3与载体强相互作用的作用位,导致在焙烧过程中WO3易发生团聚形成体相晶体,降低了WO3及Al2O3载体所提供的酸性位数量。
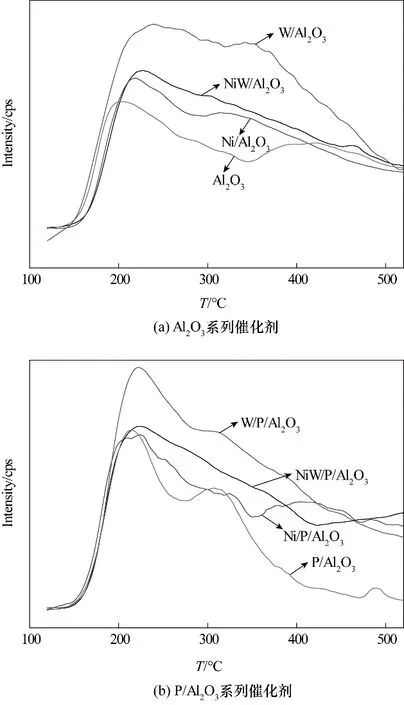
图3 Al2O3及P/Al2O3系列催化剂的NH3-TPD谱图Fig.3 NH3-TPD spectra of Al2O3 catalysts and P/Al2O3 catalysts
2.4 Al2O3及P/Al2O3系列催化剂的Py-IR表征
根据Py-IR谱图可得到如表2所示的NiW/Al2O3及NiW/P/Al2O3催化剂表面不同强度的B酸和L酸中心数量数据。根据表2 数据可以看出,NiW/P/Al2O3催化剂L 酸中心数量显著大于NiW/Al2O3催化剂L酸中心数量,从酸分布来看,磷改性后NiW/Al2O3催化剂强酸弱酸增加幅度相近,原因是磷的引入使Al2O3载体表面形成了更多的磷羟基,同时也改善了NiW 活性金属的分散,创造了更多的酸性位。催化剂的酸性对喹啉、吲哚和DBT 的吸附性能有着非常重要的作用,L酸、B酸中心数量的增加有利于喹啉、吲哚和DBT的吸附[22]。
2.5 氮化物吸附性能研究
2.5.1 氮化物在Al2O3及P/Al2O3系列催化剂上的动态吸附 图4 展示了喹啉和吲哚在Al2O3及P/Al2O3系列催化剂上的动态吸附结果,选取吸附终点时间为500 min。其中,喹啉在Al2O3系列催化剂上的吸附曲线分别记为:Q-Al2O3、Q-Ni/Al2O3、Q-W/Al2O3、Q-NiW/Al2O3;吲哚在Al2O3系列催化剂上的吸附曲线分别记为:In-Al2O3、In-Ni/Al2O3、In-W/Al2O3、In-NiW/Al2O3;喹啉在P/Al2O3系列催化剂上的吸附曲线分 别 记 为:Q-P/Al2O3、Q-Ni/P/Al2O3、Q-W/P/Al2O3、Q-NiW/P/Al2O3;吲哚在P/Al2O3系列催化剂上的吸附曲线分别记为:In-P/Al2O3、In-Ni/P/Al2O3、In-W/P/Al2O3、In-NiW/P/Al2O3。图4(a)中,喹啉在Al2O3系列催化剂上吸附的初始阶段即前80 min,其吸附量变化较大,吸附速度较快,为快速吸附阶段;当吸附时间在80~300 min 之间时,喹啉的吸附量变化缓慢,为缓慢吸附阶段;当吸附时间大于300 min 时,喹啉的吸附量变化较小,为吸附平衡阶段。同理,根据图4(b)~(c)可以看出,吲哚在Al2O3系列催化剂上的快速吸附阶段为前100 min,缓慢吸附阶段为100~400 min之间,吸附平衡阶段为400 min之后;喹啉在P/Al2O3系列催化剂上快速吸附阶段为前70 min,缓慢吸附阶段为70~350 min 之间,吸附平衡阶段为350 min 之后;吲哚在P/Al2O3系列催化剂上的快速吸附阶段为前100 min,缓慢吸附阶段为100~400 min 之间,吸附平衡阶段为400 min 之后。对比碱性氮化物喹啉与非碱性氮化物吲哚的动态吸附曲线可以发现,喹啉和吲哚在Al2O3及P/Al2O3系列催化剂的吸附状态存在较大差异,相比于吲哚,喹啉的吸附速度快、吸附量大且达到平衡所需时间短,这是因为喹啉属于碱性氮化物,其氮原子上存在孤对电子,易与Al2O3系列催化剂表面的酸性位以及金属中心结合[7]。对比喹啉与吲哚在P/Al2O3系列催化剂上的动态吸附曲线与二者在Al2O3系列催化剂上的动态吸附曲线可以发现,经磷改性后的催化剂能够在相同甚至更短的时间内吸附更多的喹啉或者吲哚,说明磷能够有效提高喹啉和吲哚在催化剂上的吸附速度,这是因为磷的引入改善了Al2O3载体的表面性质,提高了Al2O3载体表面酸中心数量,减弱了活性金属与Al2O3之间的相互作用从而提高了活性金属的堆垛层数和片晶长度,促进了氮化物的吸附[23]。

表2 Al2O3及P/Al2O3系列催化剂的酸性质Table 2 Acid property of Al2O3 catalysts and P/Al2O3 catalysts

图4 喹啉和吲哚在Al2O3及P/Al2O3系列催化剂上的动态吸附Fig.4 Dynamic adsorption of quinoline and indole on Al2O3 catalysts and P/Al2O3 catalysts
2.5.2 氮化物在Al2O3及P/Al2O3系列催化剂上的静态吸附 喹啉和吲哚在Al2O3及P/Al2O3系列催化剂上静态吸附48 h 后得到如表3 所示的平衡吸附数据。由表3 可以看出,单独负载NiO 的Al2O3载体或P/Al2O3载体对喹啉和吲哚的吸附量要小于Al2O3载体或P/Al2O3载体对喹啉和吲哚的吸附量,这是因为NiO 阻塞了Al2O3载体孔道,降低了载体的比表面积,相应地导致其吸附位较少,而NiO并没有吸附喹啉和吲哚的能力。单独负载WO3的Al2O3载体或P/Al2O3载体对喹啉和吲哚的吸附量显著大于Al2O3载体或P/Al2O3载体对喹啉和吲哚的吸附量,可见喹啉和吲哚除了在Al2O3载体上有一定的吸附能力,在活性金属WO3上也具备较强的吸附能力,且该吸附能力所带来的影响远大于比表面积下降造成的负面效应。而当Al2O3载体或P/Al2O3载体同时负载NiO和WO3时,其吸附能力略大于单独负载WO3的Al2O3载体或P/Al2O3载体,造成该现象的原因可能是Ni原子促进了WO3晶相的形成,增大了WO3上未占满分子轨道的延伸度,使其更适于对喹啉和吲哚的吸附[24]。值得注意的是,喹啉和吲哚在P/Al2O3系列催化剂上的平衡吸附量都要大于未改性催化剂上的平衡吸附量,这同样也是因为磷的引入创造了更多酸中心数量,而且通过减弱活性金属与Al2O3之间的相互作用提高了活性金属的分散度从而增大了氮化物在活性金属上的吸附比例,使更多的氮化物能够吸附在催化剂上。
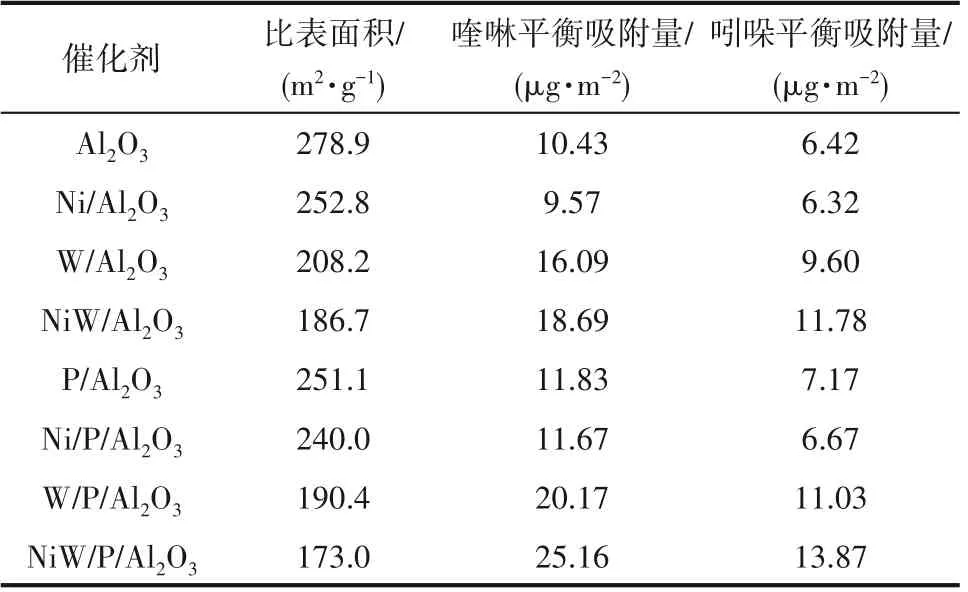
表3 喹啉和吲哚在Al2O3及P/Al2O3系列催化剂的平衡吸附量Table 3 Equilibrium adsorption capacity of quinoline and indole on Al2O3 catalysts and P/Al2O3 catalysts
2.6 硫化物吸附性能研究
2.6.1 DBT 在Al2O3及P/Al2O3系列催化剂上的动态吸附 DBT 在Al2O3及P/Al2O3系列催化剂上的动态吸附结果如图5 所示,选取吸附终点时间为500 min。其中DBT在Al2O3系列催化剂上的吸附曲线分别 记 为:DBT-Al2O3、DBT-Ni/Al2O3、DBT-W/Al2O3、DBT-NiW/Al2O3,在P/Al2O3系列催化剂上的吸附曲线分别记为:DBT-P/Al2O3、DBT-Ni/P/Al2O3、DBT-W/P/Al2O3、DBT-NiW/P/Al2O3。图5(a)中,DBT 在Al2O3系列催化剂上的快速吸附阶段为前70 min,缓慢吸附阶段为70~350 min 之间,吸附平衡阶段为350 min 之后。图5(b)中,虽然DBT 在P/Al2O3系列催化剂上动态吸附的三个阶段与未改性时相同,但是在相同时间内P/Al2O3系列催化剂对DBT 的吸附量更大,说明磷的引入对DBT 的吸附速度也有所提升,其原因与P/Al2O3系列催化剂对氮化物的吸附一致,主要体现在催化剂酸中心数量及活性金属分散度两方面。
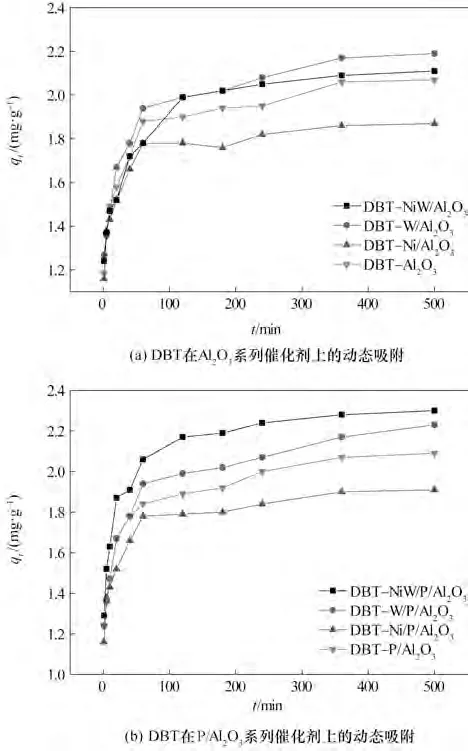
图5 DBT在Al2O3及P/Al2O3系列催化剂上的动态吸附Fig.5 Dynamic adsorption of DBT on Al2O3 catalysts and P/Al2O3 catalysts
2.6.2 DBT 在Al2O3及P/Al2O3系列催化剂上的静态吸附 表4 展示了DBT 在Al2O3以及P/Al2O3系列催化剂上静态吸附48 h 后得到的平衡吸附数据,其中DBT 饱和吸附量为催化剂比表面积与DBT 平衡吸附量的乘积,表示单位质量催化剂上DBT 的吸附质量。由表可知,DBT在NiW/Al2O3上的平衡吸附量略小于在W/Al2O3上的平衡吸附量,这可能是因为部分DBT 以平躺吸附的方式吸附在NiW/Al2O3催化剂中,导致一个DBT 分子占据了多个吸附中心,从而降低了其吸附量。除此之外,DBT 在磷改性前后Al2O3系列催化剂上的平衡吸附量与活性金属负载类型也存在以下规律,即单独负载NiO 时硫氮化合物的平衡吸附量降低,单独负载WO3时硫氮化合物的平衡吸附量得到较大程度提高,而同时负载NiO和WO3时硫氮化合物的平衡吸附量得到进一步提高[25]。
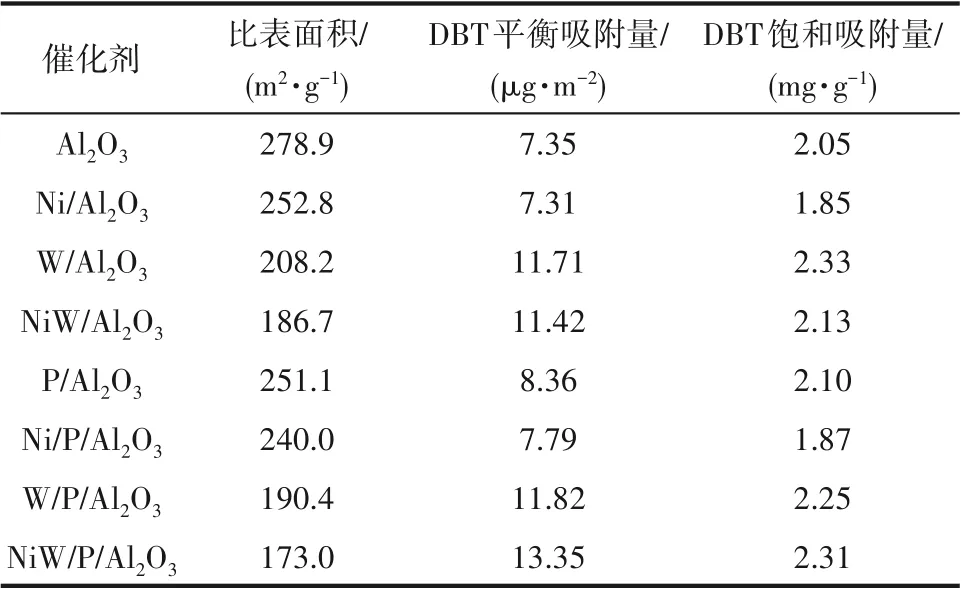
表4 DBT在Al2O3及P/Al2O3系列催化剂的平衡吸附量Table 4 Equilibrium adsorption capacity of DBT on Al2O3 catalysts and P/Al2O3 catalysts
2.7 氮化物和硫化物竞争吸附研究
2.7.1 喹啉、吲哚和DBT在催化剂上的动态吸附 喹啉、吲哚和DBT 在NiW/Al2O3以及NiW/P/Al2O3催化剂上的动态吸附结果如图6 所示,选取吸附终点时间为500 min,吸附曲线分别记为:Q-A、In-A、DBT-A 和Q-PA、In-PA、DBT-PA。当喹啉、吲哚和DBT共同存在时,在吸附的初始阶段即前50 min,吸附量变化较大,吸附速度较快,为快速吸附阶段;当吸附时间在50 ~300 min 之间时,吸附量变化较小,为缓慢吸附阶段;当吸附时间超过300 min 时,吸附量基本没有变化,为吸附平衡阶段。由吸附曲线可以看出,喹啉、吲哚和DBT 在NiW/Al2O3以及NiW/P/Al2O3催化剂上的吸附速度存在以下关系:Q-PA>QA>In-PA≈DBT-PA>In-A≈DBT-A。 不 论 在NiW/Al2O3还是NiW/P/Al2O3催化剂上,喹啉都表现出最大的竞争吸附速度,说明即使在吲哚和DBT 存在的条件下,喹啉在催化剂上的吸附仍占优势,这是因为相比于吲哚和DBT,喹啉为碱性化合物且具有较强极性,使得喹啉易于吸附在Al2O3载体表面酸性位及活性金属上。而在竞争吸附条件下,吲哚和DBT 在NiW/Al2O3催化剂或NiW/P/Al2O3催化剂上的吸附速度并无明显差异,说明吲哚和DBT 在NiWAl2O3催化剂或NiW/P/Al2O3催化剂上的吸附能力基本相同。由图6 还可发现喹啉、吲哚和DBT 在NiW/P/Al2O3催化剂上的吸附速度显著大于在NiW/Al2O3催化剂上的吸附速度,这也证明了磷的引入改变了载体表面性质及酸性,导致了吸附速度的显著变化。

图6 喹啉、吲哚及DBT在NiW/Al2O3及NiW/P/Al2O3催化剂上的动态吸附Fig.6 Dynamic adsorption of quinoline,indole and DBT on NiW/Al2O3 catalysts and NiW/P/Al2O3 catalysts
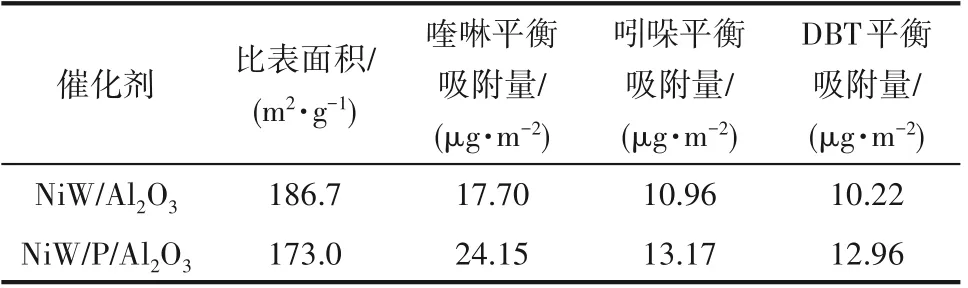
表5 喹啉、吲哚及DBT在NiW/Al2O3及NiW/P/Al2O3催化剂上的平衡吸附量Table 5 Equilibrium adsorption capacity of quinoline,indole and DBT on NiW/Al2O3 catalysts and NiW/P/Al2O3 catalysts
2.7.2 喹啉、吲哚和DBT在催化剂上的静态吸附 喹啉、吲哚和DBT 在NiW/Al2O3以及NiW/P/Al2O3催化剂上静态吸附48 h 后得到的平衡吸附数据如表5所示。由表5 可知吲哚和DBT 在NiW/Al2O3以及NiW/P/Al2O3催化剂上的平衡吸附量相接近,但明显小于喹啉的平衡吸附量,说明在竞争吸附时,喹啉仍占据了最多吸附位。对比表3 和表4 中喹啉、吲哚和DBT 单独在NiW/Al2O3以及NiW/P/Al2O3催化剂上的平衡吸附量不难发现,当三者共存时各吸附质的平衡吸附量都有所下降,这是由于三者在催化剂上的部分吸附位相同,存在竞争吸附所导致的。
2.8 氮硫化合物电荷性质与极性对吸附过程的影响

图7 喹啉、吲哚及DBT电荷分布示意图Fig.7 Scheme of charge distribution of quinoline,indole and DBT
2.8.1 喹啉、吲哚和DBT 电荷性质分析 喹啉、吲哚和DBT 在催化剂上的吸附不仅受催化剂表面性质影响,还与其本身的静电学性质密切相关。采用Gaussian 模拟方法对喹啉、吲哚和DBT 进行量子化学计算,得到如图7 所示的电荷分布。从图中可以看出,碱性氮化物喹啉氮原子上的电子云密度高具有较强的电负性,主要是因为氮原子上的孤对电子不与苯环或氮杂环上的电子云共轭,电子云主要集中在氮原子周围[26]。非碱性氮化物吲哚上的氮原子电子云密度较低,主要是因为氮原子与苯环和氮杂环发生了共轭,使氮原子上的电子云通过共轭作用向相邻的碳原子传递。而硫化物DBT 上的硫原子电子云密度最低。硫氮化合物中杂原子周围电子云密度越高,越易与酸性催化剂表面发生作用,主要体现在吸附速度的增大以及平衡吸附量的增加两方面,这也解释了喹啉、吲哚和DBT 在催化剂上表现出不同吸附性能的原因。
2.8.2 喹啉、吲哚和DBT 极性分析 化合物分子的吸附性能还与分子极性存在一定关系,而分子极性的大小可以用分子偶极矩来衡量。通过Gaussian模拟方法对喹啉、吲哚及DBT 的分子偶极矩、HOMO轨道特征值及C—N、C—S 键长进行计算分析,结果如图8 和表6 所示。由图8 和表6 可以看出,喹啉和吲哚的偶极矩显著大于DBT,故喹啉和吲哚有较强的吸附能力,能够占据催化剂上部分极性较强的活性中心。从偶极矩的方向来判断,喹啉、DBT 由于电负性较大杂原子直接暴露在分子的最外侧,因此其偶极矩的方向从杂原子一侧指向分子内部。而吲哚氮原子上连接了氢原子,主要的偶极由氮、氢这两个原子提供,因此其偶极矩方向是从氮原子指向氢原子。由此可以看出,DBT 与喹啉具有相同方向的偶极矩,其杂原子可以与催化剂的缺电子部分相互吸引。虽然DBT 硫原子电子云密度最低,但是吸附性能与吲哚并无太大差异,这是因为DBT 和喹啉都具有较强的端连吸附能力,而喹啉因为其极性更强故吸附能力也强于DBT。结合吸附数据可知,碱性氮化物喹啉在酸性催化剂表面的吸附以端连吸附为主;非碱性氮化物吲哚主要通过环上的π 键以平躺吸附的方式与催化剂酸中心或金属中心作用;DBT 在催化剂表面既发生端连吸附,又存在部分平躺吸附[27-28]。
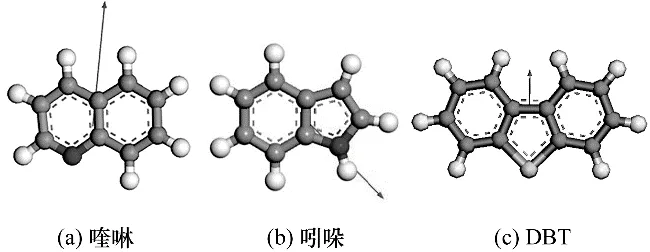
图8 喹啉、吲哚及DBT分子偶极矩示意图Fig.8 Scheme of dipole moments of quinoline,indole and DBT
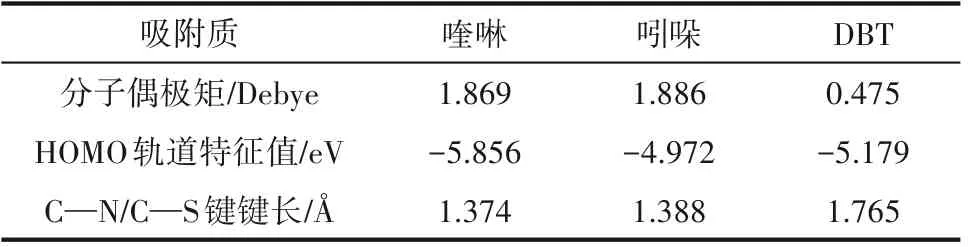
表6 喹啉、吲哚及DBT的分子结构特点Table 6 Structural characteristics of quinoline,indole and DBT
3 结 论
(1)相比于吲哚和DBT,喹啉在Al2O3及P/Al2O3系列催化剂上吸附时表现出最大的吸附速度和平衡吸附量,而吲哚和DBT 的吸附速度及平衡吸附量无显著差异。
(2)Al2O3系列催化剂经磷改性后,虽然比表面积和孔体积都有所减小,但是平均孔径得到提高,对喹啉、吲哚及DBT 的吸附速度和平衡吸附量也有较大提高,在竞争吸附条件下,NiW/Al2O3经磷改性后喹啉、吲哚及DBT 的平衡吸附量分别提高了36%、20%和27%。
(3)硫氮化合物在催化剂上的吸附不仅受催化剂表面酸性、活性金属类型以及活性金属分散度等因素影响,还与硫氮化合物分子的静电学性质与极性密切相关,催化剂表面酸性越强或酸中心数量越多、活性金属分散度越高、硫氮化合物分子杂原子电子云密度越大或分子极性越大,硫氮化合物在催化剂上的吸附速度和平衡吸附量越大。
