4-氟吡啶-2-甲腈合成工艺研究
2021-04-08张成新冯伟李远超李善清孟烨
张成新,冯伟,李远超,李善清,孟烨
(1.山东省海洋精细化工重点实验室,山东 潍坊 262737;2.山东省海洋化工科学研究院,山东 潍坊 262737)
4-氟吡啶-2-甲腈是一种重要的医药和化工中间体[1-3]。文献报道的4-氟吡啶-2-甲腈合成方法主要是以4-硝基吡啶-2-甲腈、四丁基氟化铵为原料,经氟代脱硝反应制得[1-4]。氟代脱硝法是一种在目标物分子上引入氟原子的常用方法[2-7]。该法存在以下不足:1)原料需提前在特定位置引入硝基,制备工艺难度大;2)芳环化合物参与的氟代脱硝反应易生成酚和醚类杂质;3)氟代脱硝反应收率受四丁基氟化铵的水含量影响较大,无水四丁基氟化铵制备工艺苛刻;4)四丁基氟化铵的相对分子质量较大,作为氟代试剂用量大、回收处理难度大。近年来,通过卤素交换反应在目标分子上引入氟原子已成为制备含氟有机化合物的另一种有效方法[8-10],使用卤素交换方法制备含氟有机化合物成本相对较低,更有利于工业化生产。4-氟吡啶-2-甲腈合成方法存在原料4-硝基吡啶-2-甲腈价格昂贵,无商业化产品,不易获得;氟代试剂无水四丁基氟化铵制备工艺苛刻,价格昂贵等缺陷,无法满足规模化生产的要求,而通过卤素交换制备4-氟吡啶-2-甲腈的方法尚无文献报道。鉴于此,本文开发了以4-氯吡啶-2-甲酸甲酯为原料,经氨解反应、酰胺脱水反应、卤素交换反应制备4-氟吡啶-2-甲腈的合成路线。该合成路线工艺条件温和、原料成本低、适合工业化生产,具体合成路线如图1所示。
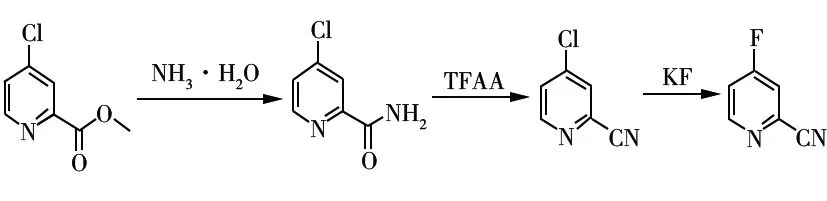
图1 4-氟吡啶-2-甲腈的合成路线
1 实验部分
1.1 仪器与试剂
Bruker Avance 500 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标),瑞士Bruker公司;7890A型气相色谱仪(GC),美国Agilent公司;PE2400元素分析仪,美国PerkinElmer公司;TGA/DSC1/1100LF热重/差热同步热分析仪,瑞士METTLER TOLEDO公司;Clarus 690 SQ8气相色谱-质谱联用仪,美国PerkinElmer公司;IRPrestige-21红外光谱仪,日本岛津公司。4-氯吡啶-2-甲酸甲酯,工业级≥98%,GC检测,上海毕得医药科技有限公司;无水氟化钾,工业级≥99.5%,检测方法GB/T 28652—2012,新乡市金沙化工有限公司;其余所用试剂均为国产分析纯。气相色谱检测方法:柱子HP-5(30 m×0.32 mm,膜厚0.25 μm),高纯氮气,1.5 mL/min,柱温60 ℃保持2 min;8 ℃/min升至260 ℃保持3 min,检测器温度300 ℃。
1.2 合 成
1.2.1 4-氯吡啶-2-甲酰胺的合成[11]
在1 L反应瓶中加入85.8 g(0.50 mol) 4-氯吡啶-2-甲酸甲酯、430 g甲醇,搅拌溶解。20~30 ℃滴加84.1 g质量分数为25%~28%的氨水,滴加时间约1 h。滴加完毕继续在20~30 ℃下搅拌反应6~8 h。反应结束,降温至0~5 ℃析晶0.5~1 h,过滤得到4-氯吡啶-2-甲酰胺。m.p. 161.3~162.6 ℃,MS(EI)m/z:156.0[M+],GC纯度99.2%,收率91.6%。滤液中和后蒸馏回收甲醇,剩余部分进高浓度废水处理系统。
1.2.2 4-氯吡啶-2-甲腈的合成[11-12]
在1 L反应瓶中依次加入62.6 g(0.40 mol) 4-氯吡啶-2-甲酰胺,81.0 g(0.80 mol)三乙胺,470 g四氢呋喃,搅拌溶解。置于低温浴槽中降温至-10~0 ℃,滴加92.4 g(0.44 mol)三氟乙酸酐(TFAA),约1 h滴毕。-10~0 ℃保温反应1 h,然后升温至30~35 ℃保温反应3 h。反应结束,减压蒸馏除去四氢呋喃,加入150 g水和200 g乙酸乙酯,分液、水洗,收集有机相,减压蒸馏除去乙酸乙酯得到4-氯吡啶-2-甲腈。m.p. 83.3~84.8 ℃,MS(EI)m/z:138.0[M+],GC纯度98.3%,收率86.3%。水洗废水中和后进高浓度废水处理系统。
1.2.3 4-氟吡啶-2-甲腈的合成[13-15]
在500 mL反应瓶中加入49.9 g(0.36 mol) 4-氯吡啶-2-甲腈,11.6 g(0.036 mol)四丁基溴化铵,41.8 g(0.72 mol) 粉末状干燥氟化钾,216 gN,N′-二甲基甲酰胺,氮气保护,升温至130~140 ℃反应3 h。反应完毕,降至室温,过滤除去反应体系中的盐,滤液加入648 g水和320 g二氯甲烷,分液、水洗,收集有机相,减压蒸馏除去二氯甲烷,加入60 g水,搅拌析晶,过滤得到4-氟吡啶-2-甲腈。m.p. 74.7~77.1 ℃,MS(EI)m/z:122.0[M+],GC纯度99.2%,收率83.7%。水洗废水进入焚烧废水处理系统,析晶滤液进入高浓度废水处理系统。
2 结果与讨论
2.1 4-氯吡啶-2-甲酰胺的合成条件优化
该步反应为酯的氨解反应,属于亲核取代反应。由于受到吡啶环吸电子作用影响,酯基非常活泼,酯氨解反应在20~30 ℃条件下即可发生。氨水的用量是该步反应的关键影响因素,在保持其他反应条件不变的情况下,考察了氨水用量对反应收率的影响,见表1。从表1中可以看出,随着氨水用量(w=25%~28%)的增加,4-氯吡啶-2-甲酰胺的收率呈现先增加后减小的趋势。当氨水用量从0.48~0.54 mol逐渐增大到0.6~0.67 mol时,4-氯吡啶-2-甲酰胺收率不断增加并达到峰值;继续增加氨水用量,体系碱性增强,4-氯吡啶-2-甲酸甲酯转化成4-氯吡啶-2-甲酰胺后会进一步发生水解生成4-氯吡啶-2-甲酸并与氨水成盐,导致4-氯吡啶-2-甲酰胺收率降低。当n(氨水)∶n(4-氯吡啶-2-甲酸甲酯)为1.2~1.34∶1时,4-氯吡啶-2-甲酰胺的收率最高,反应结果最佳。

表1 氨水用量对4-氯吡啶-2-甲酰胺收率的影响
2.2 4-氯吡啶-2-甲腈的合成条件优化
酰胺脱水制备腈是经典反应,本文选择三氟乙酸酐为脱水剂,三乙胺为缚酸剂制备4-氯吡啶-2-甲腈。三氟乙酸酐的用量是影响4-氯吡啶-2-甲腈收率的关键因素,实验结果见表2。由表2可知,当n(三氟乙酸酐)∶n(4-氯吡啶-2-甲酰胺)从1∶1增加到1.1∶1时,产物收率从80.6%增加到86.3%,继续增大三氟乙酸酐与4-氯吡啶-2-甲酰胺的摩尔比,产物收率无明显增加,基本稳定在86.3%。因此当n(三氟乙酸酐)∶n(4-氯吡啶-2-甲酰胺)为1.1∶1时,产物收率最高,反应结果最佳。

表2 物料比对4-氯吡啶-2-甲腈收率的影响
2.3 4-氟吡啶-2-甲腈的合成条件优化
卤素交换反应一般是指在极性非质子溶剂中,碱金属氟化物或有机氟化物等氟源与反应底物分子中的卤素原子之间进行的SN2型亲核取代反应,其中应用最广的是氟氯交换反应。为了降低原料成本,本文采用价格低廉的氟化钾为氟源。但氟化钾为无机盐,在有机溶剂中的溶解度非常小,因此需要在反应体系中添加相转移催化剂提升固液两相间的传质能力,加快反应速度。卤素交换氟代反应催化剂主要有季鏻盐、季铵盐、冠醚及其2种或3种的复配催化剂等,其中季铵盐应用最广。由表3可知,不加催化剂卤素交换氟代产物收率较低,反应结果较差;加入催化剂后卤素交换氟代反应收率明显提高,其中以四丁基溴化铵为催化剂时,反应结果最佳。

表3 催化剂对4-氟吡啶-2-甲腈收率的影响
卤素交换反应为亲核取代反应机理,溶剂必须为极性非质子溶剂。在其他反应条件不变情况下,考察了不同溶剂和反应时间对反应收率的影响,见表4。从表4可知,N,N′-二甲基甲酰胺、二甲基亚砜、N-甲基吡咯烷酮、环丁砜为反应溶剂时,反应收率都较好,其中以N,N′-二甲基甲酰胺为反应溶剂时,反应效果最佳,收率最高。从表4中的反应时间对反应收率影响可以看出,反应时间为3 h或5 h,收率差异不大,因此较佳的反应时间为3 h。

表4 溶剂对4-氟吡啶-2-甲腈收率的影响
2.4 目标化合物的结构表征
由目标化合物的热重分析(TGA)和差热扫描量热(DSC)谱图可知,目标化合物的熔点(Tm)为77.1 ℃,由于产物易升华,5%热失重仅为85.5 ℃。
FT-IR(KBr):3 087、3 061 cm-1是吡啶环上的C—H伸缩振动吸收峰,1 583、1 467、1 401 cm-1是不饱和双键C=N,C=C伸缩振动吸收峰,2 242 cm-1是C≡N的伸缩振动吸收峰;1 273 cm-1是C—F的伸缩振动吸收峰。
1H NMR (500 MHz,CDCl3),δ:8.72~8.75(dd, 1H),7.51~7.53 (dd, 1H),7.33~7.36(m, 1H)。元素分析C6H3FN2(%):计算值C 59.02, H 2.48, N 22.94;测试值C 59.0, H 2.51, N 22.93;质谱分析MS(EI)m/z: 122.0[M+],与理论计算一致。
3 结 论
以4-氯吡啶-2-甲酸甲酯为原料,经氨解反应、酰胺脱水、卤素交换反应得到目标化合物4-氟吡啶-2-甲腈,并对每步反应的关键影响因素作了优化考察。该方法具有反应路线短、操作简单、原材料成本低、产品纯度高等优点,为化合物4-氟吡啶-2-甲腈的制备提供了一种简便有效的合成方法。
