因复发性胎儿心脏畸形而确诊的一例22q11.21微缺失家系
2021-04-06李丽萍邹永毅肖菊花刘艳秋
李丽萍,邹永毅,肖菊花,刘艳秋
(1.江西省妇幼保健院产前诊断中心,江西省出生缺陷防控重点实验室;2.江西省妇幼保健院超声诊断科,江西 南昌 330006)
22q11.2微缺失综合征(22q11.2 deletion Syndrom,22q11.2 DS)是常见的由于22q11.2区域(22q11.21-22q11.23)拷贝数变异(copy number variations,CNVs)引起的染色体微缺失综合征[1]。近年来,随着分子检测技术的进步和产前诊断病例的积累,22q11.2微缺失胎儿的患病率约为1/1000,如有超声结构异常,特别是有先天性心脏病的胎儿,患病率可高达1/100[2-3]。22q11.2 DS的临床表现多种多样并且轻重程度不一。主要表现有先天性心脏病、发育迟缓、免疫缺陷和特殊面容、内分泌异常,肾脏发育不全,行为问题和精神异常[1,4]。本研究采用染色体核型分析和染色体微阵列分析(CMA)技术对1例复发性胎儿心脏畸形孕史的患者进行细胞遗传学及分子遗传学检测,明确其染色体拷贝数变异的性质,探讨其发病机制及预后,以期达到对22q11.2 DS家庭的遗传咨询提供一些指导及建议,报告如下。
1 资料与方法
1.1 一般资料 2019年11月13日江西省妇幼保健院就诊患者1例,因“胎儿心脏畸形孕史2次”至产前诊断中心进行遗传咨询。患者年龄29岁,大专学历,孕2产0,曾孕2次均因“胎儿室间隔缺损,永存动脉干”引产,(第2次胎儿“室缺,永存动脉干”超声结果图像见图1)。非近亲结婚,否认有遗传病家族史,否认有已知有毒有害物质接触史,青霉素、头孢过敏史。由于两次引产胎儿均未行相关遗传学检查,因此为明确胎儿心脏畸形的病因,对其进行常规G显带染色体核型分析和CMA检测。
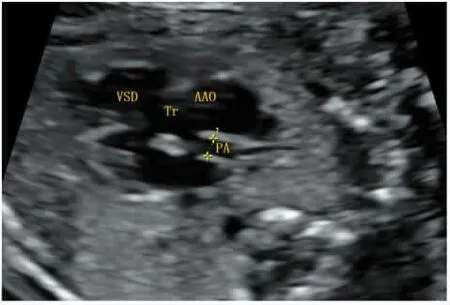
图1 流出道切面显示室间隔回声中断(VSD),仅见一组半月瓣,粗大动脉干(Tr)骑跨在室间隔上,动脉干在半月瓣稍上方分出升主动脉(AAO)和肺动脉(PA)
1.2 方法
1.2.1 外周血染色体核型分析 抽取患者外周血2 ml,接种约1ml全血于淋巴细胞培养液中(达辉生物技术公司),37℃,5%二氧化碳培养箱中进行培养72h。培养终止前4h加入秋水仙素(20μg/ml),按常规方法制片,G显带,分辨率为320~400条带。使用莱卡染色体核型扫描分析系统,分析20个分裂相。核型分析依据人类细胞遗传学国际命名体制(ISCN2016)标准进行。
1.2.2 染色体微阵列分析技术 采用QIA-GEN公司生产的Qiamp DNA Blood Mini Kit进行外周血DNA提取,使用Affymetrix Cytoscan 750K(美国Affymetrix公司)芯片进行拷贝数变异检测,操作步骤严格按照说明进行,由Affymetrix@Chromosome Analysis Suite 2.0(ChAS2.0)软件对芯片进行数据扫描和分析。
2 结果
2.1 外周血细胞染色体核型分析结果 G显带细胞染色体核型未见异常,核型结果为46,XX,见图2。

图2 G显带细胞染色体核型图
2.2 染色体微阵列分析结果 患者的CMA分析结果由上海交通大学医学院附属国际和平妇幼保健院出具,其显示arr[GRCh37]22q11.21(18648855-21800471)×1,即患者的22号染色体长臂11.21区域发现3.15Mb的缺失,其他区域未见拷贝数目异常,该缺失区间与DECIPHER收录的综合征22q11 deletion syndrome基本重叠,该区域包含OMIM数据库中功能基因TBX1(OMIM188400)。该基因与22q11.2 DS的五大主要表型(异常面容、心脏缺陷、胸腺发育不良、腭裂、甲状旁腺功能不全与低钙血症)相关[5]。见图3。
2.3 家庭成员的染色体微阵列分析结果 为了追溯患者22q11.21微缺失的起源,对其父母及丈夫进行CMA检测。结果显示其母亲及丈夫基因组DNA拷贝数目未见异常,然而其父亲CMA结果显示在相同位置arr[GRCh37]22q11.21(18648855-21800471)发现3.15Mb缺失,见图3。

图3 患者染色体微阵列分析信号图,红色箭头指示染色体22q11.21缺失区域,红圈为缺失区域的放大图
2.4 该患者的一些临床特征 身高中等(1.56m),长脸,小嘴,小下颌,低耳位,在与其沟通中发现其讲话口齿不清,鼻音重,但无明显唇腭裂。她没有行为或精神问题,没有频繁呼吸道感染或癫痫病史。于南昌大学第二附属医院行心脏彩超提示:二尖瓣、三尖瓣、主动脉瓣轻度反流,其余未见异常。这些检查结果显示,患者有轻微异常,但没有明显临床问题。
3 讨论
22q11.2微缺失是指染色体22q11.2区域发生的0.7~3 Mb的片段缺失。由于缺失片段小,传统的G显带染色体核型分析方法分辨率较低,目前已不适宜运用于22q11.2微缺失的临床检测[6]。另有分子生物学技术如荧光原位杂交,也能够对22q11.2微缺失进行检测,但由于其检测探针较少,可能导致不能全面覆盖22q11.2的微缺失,造成漏检,同时其通量较低,难以大批量检测[6-7]。随着遗学研究方法的发展,染色体微阵列分析技术(CMA)作为一种新型的分子诊断方法逐渐应用起来。该技术具有通量高、分辨率高,可在全基因组范围内检测等优点[8-9]。
染色体22q11.2区域在减数分裂时易发生染色体重排,导致先天性畸形综合征,其中最典型的是22q11.2微缺失综合征(22q11.2 DS)。有研究报道显示此类综合征仅有5%是遗传性的[1,10]。如果该缺失综合征为家族性遗传,则符合常染色体显性遗传模式[11]。本案例中患者与其父亲的22q11.21相同位置均发现3.15Mb缺失,说明患者该区域的缺失遗传自其父亲,符合常染色体显性遗传规律。因患者的爷爷奶奶均去世,故无法追溯患者父亲22q11.2微缺失的来源。
22q11.2 DS临床表现多样、复杂,表型和基因型之间关联不密切,这给22q11.2 DS的临床诊断带来了困难[12]。心脏异常往往是22q11.2微缺失最主要的表现,据报道约74%的病例患有先天性心脏病,尤以圆锥动脉干畸形最为常见,其包括法洛四联症,主动脉弓中断,室间隔缺损、永存动脉干及右室双出口[1,13]。这些复杂先心病治疗困难、新生儿手术非常危险,从而导致该病的病死率非常高,预后也极差。但本报道中,尽管患者及其父亲均检测出22q11.2经典微缺失,但两人均无22q11.2 DS经典的表型。2019年有学者报道在一个复发性胎儿圆锥动脉干畸形家系中发现罕见的嵌合型22q11.2微缺失[14]。另有研究报道1例因学习困难而确诊的22q11.2微缺失综合征,该患者无典型临床表现,且细胞和体液免疫检测未见明显缺陷[15]。所以在22q11.2缺失的诊断中要重视临床病史的采集,了解其临床表现的多样性,不要忽视了不明显的异常特征。
先天性心脏病的发病机制目前仍不明确,目前的研究认为遗传与环境因素相互作用是先心病的主要原因[16]。在遗传病因研究中多认为其中染色体数目或结构的异常是其主要致病原因[17]。染色体数目异常主要包括21-三体、18-三体等非整倍体变异[18]。染色体结构异常主要包括最常见的22q11微缺失、8q23缺失及7q11缺失等的拷贝数变异。本报道中该患者所孕2次胎儿均表现为22q11.2 DS常见的心脏圆锥动脉干畸形,但因两次引产胎儿均未行相关遗传学检查,所以不能完全确定胎儿心脏畸形为22q11.2缺失导致,但因患者及其父亲均检测出22q11.2缺失,所以推断胎儿为22q11.2缺失的可能性极大。又因为22q11.2缺失符合常染色体显性遗传规律,所以患者再次生育有50%的概率生育22q11.2微缺失胎儿,建议下次生育可借助辅助生殖技术行胚胎植入前检测(PGS),避免胎儿大月份引产对患者造成的生理及心理上的伤害。
因此,对于疑似妊娠22q11.2微缺失胎儿的高危孕产妇如面容畸形患者或已生育过先心儿的患者,以及结合产前超声怀疑孕育先天性心脏病或唇裂胎儿的患者,同时对于同本报道中发现的家族遗传性的22q11.2 DS患者,均需要进行产前22q11.2微缺失的检测,做到早发现、早干预及对再发风险的评估,这对于降低出生缺陷、提高人口素质具有重要意义,最终达到优生优育的目的。
