超高效液相色谱-串联质谱法测定茶叶和茶汤中茚虫威对映体及7种降解产物
2021-04-02黎洪霞罗逢健陈宗懋张新忠
钟 青,黎洪霞,罗逢健,陈宗懋,张新忠
(1.中国农业科学院茶叶研究所,农产品质量安全研究中心,浙江 杭州 310008;2.中国农业科学院研究生院, 北京 100081; 3.农业农村部茶叶质量安全控制重点实验室,浙江 杭州 310008)
我国是茶叶的起源地,也是世界第一茶叶生产国和消费国。茶叶是一种绿色健康饮料,其品质和质量安全关系着茶产业的长期可持续发展。茶叶生长喜湿热环境,容易发生病虫害,喷施化学农药是防治茶树病虫害最快、最有效的方法。茚虫威结构独特新颖、蒸气压低(2.5×10-5mPa,25 ℃)、水溶解度小(0.20 mg/L,20 ℃)、杀虫效果好且用量低,是一种对茶树鳞翅目害虫假眼小绿叶蝉等有很好的防治效果,且对天敌安全的噁二嗪类手性结构农药[1]。茚虫威通过在昆虫体内代谢为一种N-去甲氧羰基代谢物(DCJW,IN-JT333),从而不可逆地阻断Na+通道,逐渐使目标害虫神经麻痹,影响摄食等活动直至死亡[2]。2013年我国对茶叶中茚虫威的最大残留限量(MRL)规定为5 mg/kg,并促成国际食品法典委员会CAC制定此MRL值,使欧盟的MRL值从0.05 mg/kg修订为5 mg/kg。近年来提倡使用茶园水溶性农药,茚虫威作为有机磷类高毒农药、新烟碱类高水溶解度农药的替代品,越来越广泛地用于茶园害虫的防治[1,3-4]。
随着科学研究的不断深入,农产品和食品中农药代谢降解产物受到关注。国际农药残留联席会议JMPR的评估报告[5]将茚虫威残留物定义为茚虫威-S体和茚虫威-R体的总量,同时给出了一些代谢降解产物,除IN-JT333外,还有羟基化代谢物、噁二嗪开环代谢物等[6]。关于茚虫威残留的研究,前期主要集中在其外消旋体的检测方法,包括气相色谱法[7-9]、气相色谱-串联质谱法[10-12]、液相色谱法[13-14]和液相色谱-串联质谱法[15-22]等。2007年以来,逐渐有采用不同固定相制成的色谱柱对不同样品基质中的茚虫威对映体进行手性分析的报道[23-26],如采用万古霉素晶体代谢物柱[27]、(S)-α-甲基苯基氨基甲酸修饰的直链淀粉柱[28]、直链淀粉-三-(3,5-二甲基苯基氨基甲酸酯)(Chiralpak AD-RH)柱[29]和纤维素-三-(3,5-二甲基苯基氨基甲酸酯)(Chiralcel OD-H)柱[30]等。本课题组前期建立了茶叶等基质中茚虫威对映体的残留分析方法[23],并用于田间实际样品分析,发现茶鲜叶中茚虫威对映体之间存在降解差异[26]。但这些研究均只关注茚虫威母体,很少涉及其降解产物。
本工作拟采用超高效液相色谱-三重四极杆串联质谱(UPLC-MS/MS)法分析鲜叶、绿茶、红茶、绿茶茶汤和红茶茶汤中茚虫威手性对映体及其7种降解产物,希望为研究茚虫威对映体在茶叶生长加工冲泡过程中的降解代谢及在其他农产品中的代谢分析提供方法参考。
1 实验部分
1.1 主要仪器与设备
UPLC/Quattra Premier XE超高效液相色谱-三重四极杆质谱联用仪:美国Waters公司产品,配有电喷雾电离(ESI)源,MassLynx 4.1质谱工作站;Lux®3 μm Cellulose-1、Lux®3 μm Cellulose-2(3 μm×150 mm×2 mm)手性色谱柱:Strata-X 33 u PRP (polymeric reversed phase)SPE柱(500 mg/6 mL):美国Phenomenex公司产品;BondElut C18 SPE柱(500 mg/6 mL):美国Agilent公司产品;3K-5冷冻高速离心机:德国Sigma公司产品;R-210旋转蒸发仪:瑞士BUCHI Labortechnik A G公司产品;KQ-250DB型数控超声波清洗器:昆山市超声仪器有限公司产品;Vortex Genie2型涡旋振荡器:美国Scientific公司产品;T-18高速均质匀浆器:德国IKA公司产品;DFT-200手提式高速万能粉碎机:浙江温岭市林大机械有限公司产品;电子分析天平(0.000 1 g):瑞士Mettler-Toledo公司产品;Filter Unit滤膜(0.22 μm):天津博纳艾杰尔科技有限公司产品;2 mL进样瓶:美国Agilent公司有限产品。
1.2 材料与试剂
无水硫酸镁、氯化钠:均为分析纯,上海试四赫维化工有限公司产品;石墨化炭黑(Graphitized carbon black,GCB)填料(120~140目):天津博纳艾杰尔科技有限公司产品;十八烷基键合硅胶(Octadecylsilane, C18)填料:40~60 μm,天津博纳艾杰尔科技有限公司产品;丙酮:色谱纯,美国Honey Well公司产品;甲醇:色谱纯,德国Merck公司产品;甲酸、乙酸铵:色谱纯,上海安普实验科技股份有限公司产品;纯净水:杭州娃哈哈有限公司产品;茚虫威标准品(R∶S=1∶1,纯度98%):德国Dr. Ehrenstorfer公司产品;IN-JT333、IN-MK638、IN-MF014、IN-KG433、IN-MN470、IN-MK643和IN-JU873纯品:纯度均大于95%,委托合成制备。
1.3 样品提取净化
1.3.1鲜叶、茶叶 称取经食品粉碎机磨碎后的5.00 g鲜叶(或2.00 g绿茶或红茶)于50 mL离心管中,加入5 mL纯净水,充分涡旋混匀后静置20 min,再加入10 mL乙腈,涡旋混匀后静置2 h,然后加入5 g NaCl,以17 400 r/min均质1 min,涡旋混匀振荡5 min,以5 000 r/min离心5 min。吸取7 mL上层有机相溶液,加入装有0.525 g C18和0.087 5 g GCB的10 mL离心管中,涡旋振荡净化1 min,以5 000 r/min离心5 min,吸取5.0 mL过膜后的上清液于50 mL鸡心瓶中,旋转浓缩近干后,加入1.0 mL甲醇-水溶液(9∶1,V/V)定容,超声辅助溶解,过0.22 μm滤膜至进样瓶中,待UPLC-MS/MS测定。
1.3.2茶汤 按照茶水比1∶50的标准用沸水冲泡茶叶,10 min后滤纸抽滤,获得茶汤。先用5 mL甲醇、5 mL水依次预淋洗活化PRP柱,吸取100 mL茶汤样品上样,待过柱完毕后,加入5 mL甲醇-水溶液(4∶6,V/V)清洗PRP柱,弃去流出的上样液和清洗液,缓速气流吹干PRP柱2 min,再用20 mL甲醇洗脱柱内待测目标物,接收洗脱液至100 mL鸡心瓶中,旋转蒸发浓缩近干后,加入1.0 mL甲醇-水溶液(9∶1,V/V)定容,超声辅助溶解,过0.22 μm滤膜,待UPLC-MS/MS测定。
1.4 实验条件
1.4.1色谱条件 Lux®3 μm Cellulose-2色谱柱(3 μm×150 mm×2 mm);柱温40 ℃;进样量5 μL;流速0.30 mL/min;流动相:A为0.1%甲酸-甲醇溶液,B为5 mmol/L乙酸铵水溶液;梯度洗脱程序:0~4.5 min(70%~80%A),4.5~5.2 min(80%~98%A),5.2~9.2 min(98%A),9.2~9.4 min(98%~70%A),9.4%~12 min(70%A)。茚虫威对映体及其降解产物的保留时间列于表1。
1.4.2质谱条件 电喷雾电离正离子多反应监测模式(ESI+-MRM);毛细管电压3.5 kV;离子源温度150 ℃;脱溶剂气(N2)温度350 ℃,流速750 L/h;锥孔反吹气(N2)流速50 L/h;碰撞气(Ar)流速0.25 mL/min;电子倍增器电压700 V;二级母离子驻留时间0.040 s。茚虫威对映体及其降解产物的二级质谱参数列于表1。
1.5 标准溶液的配制与标准曲线
分别称取一定量的茚虫威及其降解产物标准品于50 mL容量瓶中,用乙腈溶解定容,配制成200 mg/L的标准储备液,-18 ℃保存。将标准储备液用甲醇-水溶液(9∶1,V/V)稀释成20 mg/L混合标准溶液,然后用1.3节方法处理后得到的茶鲜叶、红茶、绿茶、红茶茶汤和绿茶茶汤基质以及甲醇-水(9∶1,V/V)溶剂配制成10、5.0、2.50、1.0、0.50、0.25、0.10、0.050、0.025和0.01 mg/L系列标准溶液(对映单体浓度为一半),UPLC-MS/MS分析。每个浓度测定3次,以浓度为横坐标(x),峰面积平均值为纵坐标(y),得到茚虫威对映体及降解产物的标准曲线和线性相关系数。
基质效应(matrix effect):
ME=(A/B-1)× 100%
式中:A为基质标准曲线的斜率,B为溶剂标准曲线的斜率。若ME大于0,说明存在基质增强效应;若ME小于0,说明存在基质减弱效应。
1.6 添加回收率、精密度与方法定量限
称取(量取)经测定不含茚虫威及其降解产物的茶鲜叶、绿茶、红茶、绿茶茶汤、红茶茶汤空白样品,分别添加0.005、0.050、0.50 mg/kg 3个浓度水平的混合标准溶液,涡旋混匀后放置2 h以更接近实际样品中农药残留情况,然后按照1.3节方法加入水和乙腈进行提取净化,每个浓度重复6次;同时将处理得到的空白茶鲜叶、绿茶、红茶、绿茶汤和红茶汤样品溶液加入相应浓度的标准溶液后定容,配制成相应的基质标准溶液,计算添加回收率、相对标准偏差、检出限和定量限。
2 结果与讨论
2.1 前处理条件的优化
2.1.1茶鲜叶和茶叶提取条件的优化 提取溶剂的酸碱性直接影响样品中残留化合物的提取效果。参考前期方法[22,26]进行提取时,发现IN-JT333等化合物的回收率无法满足需要。因此,分别称取5.0 g茶鲜叶,加入1.0 mL 1 mg/L茚虫威及其降解产物混合标样,涡旋静置1 h,对比4种提取方式的提取效果,即A:加入5 mL 2%甲酸-水溶液,涡旋浸泡20 min,再加入10 mL 5%氨水-乙腈提取;B:加入5 mL纯水,涡旋浸泡20 min,再加入10 mL 5%氨水-乙腈提取;C:加入5 mL纯水,涡旋浸泡20 min,再加入10 mL乙腈提取;D:加入5 mL纯水,涡旋浸泡20 min,再加入10 mL 2%甲酸-乙腈提取。按照1.3节方法净化后,各化合物的提取回收率示于图1。结果表明,虽然5%氨水-乙腈提取液相对更干净,但对IN-JT333、IN-JU873和INKG433的回收率较低;乙腈和酸化乙腈都能得到较好的回收率,但是酸化乙腈提取重复间偏差较大,因此选择方案C。本实验最终采用5.0 mL水和10.0 mL乙腈提取5.0 g茶鲜叶,茚虫威对映体及其降解产物的回收率和精密度均能满足要求,同时也适用于茶叶的提取。
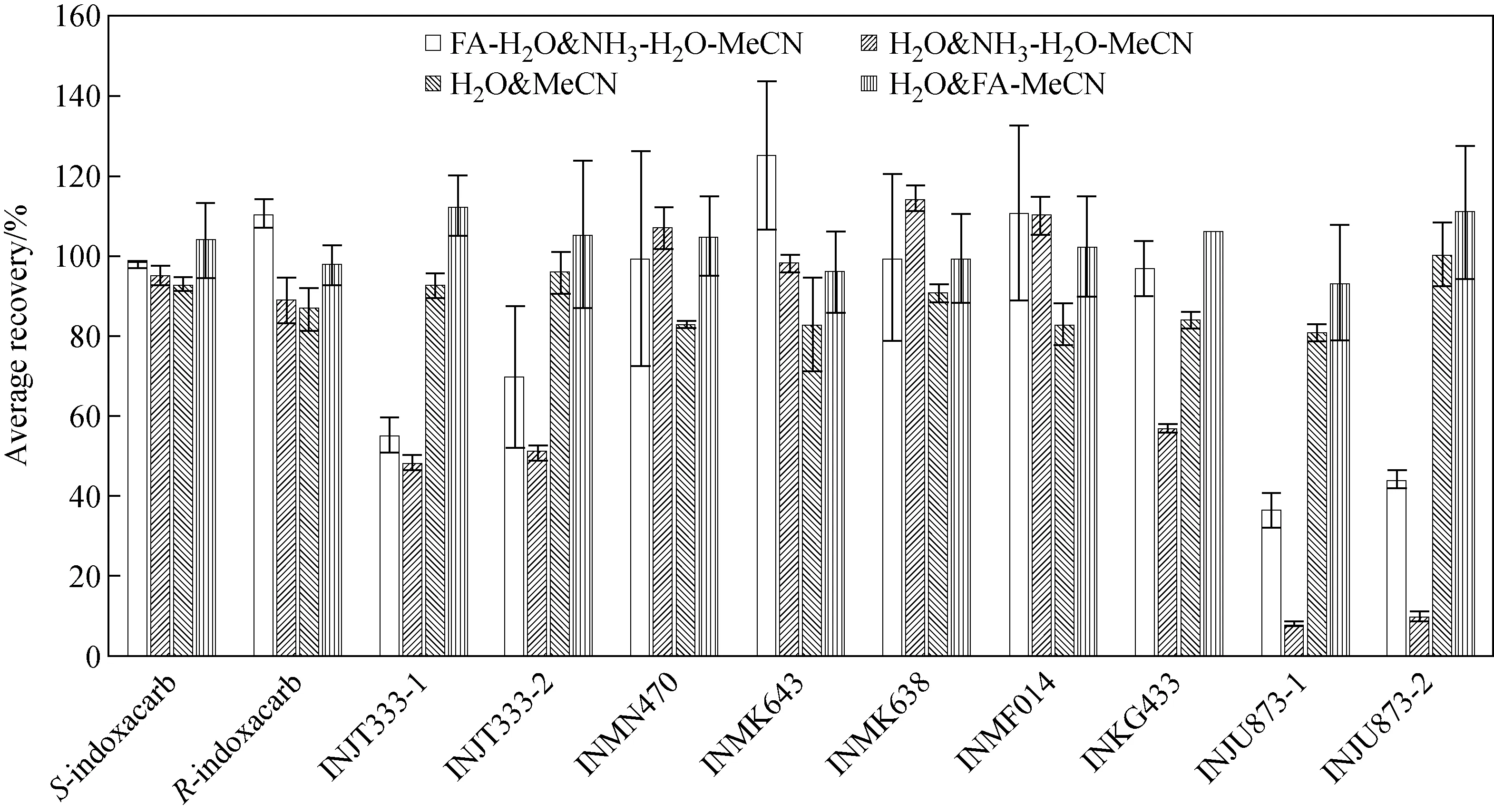
图1 4种不同提取溶剂提取鲜叶中茚虫威及其降解产物的对比结果Fig.1 Comparation results to indoxacarb and its metabolites extracted by four kinds of extractants
2.1.2茶汤富集净化条件的优化 参考前期方法[22]选择固相萃取柱和上样量,选择茚虫威、IN-JT333和IN-MK638三种极性不同的化合物,采用Bond-Elut C18-SPE柱和PRP-SPE柱,上样25、50、100 mL茶汤进行对比研究,回收率结果列于表2。虽然C18柱对茚虫威的保留富集较好,但是对IN-JT333和IN-MK638的保留富集较差,尤其是IN-MK638的回收率无法满足要求。从上样量变化带来的结果看,C18柱吸附能力不足。PRP柱采用高聚合物填料,能够增强对IN-MK638等弱极性化合物的吸附富集,从而提高回收率,满足残留分析要求。因此,最终选择PRP柱对100 mL茶汤中的茚虫威及其降解产物进行富集净化,最大限度提高富集倍数和方法灵敏度。
2.1.3PRP柱净化洗脱溶剂的优化 将茶汤样品上样至PRP柱之后,柱内吸附较多咖啡碱、色素等杂质,如果上样后采用甲醇直接洗脱,会带来很多共流出杂质,影响待测物响应。因此在不影响目标物质的情况下,将一部分咖啡碱、色素等杂质先淋洗下来,再洗脱目标化合物,从而降低基质效应,减少杂质影响。将茚虫威、IN-JT333和IN-MK638的混合标准溶液、茶汤分别上样至PRP柱,依次吸取5 mL不同比例(0∶10、1∶9、2∶8、3∶7、4∶6、5∶5、6∶4、7∶3、8∶2、9∶1、10∶0,V/V)的甲醇-水溶液进行洗脱,测定过柱后各部分洗脱液中的目标物浓度,绘制洗脱曲线,示于图2。茶汤过柱后在不同比例洗脱溶剂下的颜色对比示于图3。结果表明,大部分水溶性色素在前期水比例较高的淋洗中被洗脱下来;随着甲醇比例的升高,IN-MK638最先被洗脱下来,当甲醇-水溶液比例上升至5∶5时开始有检出,在6∶4时全部流出,之后在8∶2开始有茚虫威洗脱,最后是IN-JT333被洗脱下来,在纯甲醇时被全部洗脱下来。因此,既要尽量去除样品中咖啡碱、色素等杂质,又要避免目标物损失,最终确定上样后最佳清洗溶剂为5 mL甲醇-水溶液(4∶6,V/V),然后用甲醇洗脱待测目标化合物。

表2 不同SPE柱和不同上样量时,3种代表性化合物的回收率Table 2 Recoveries of indoxacarb, IN-JT333, IN-MK638 with different SPE columns and different sample volumes
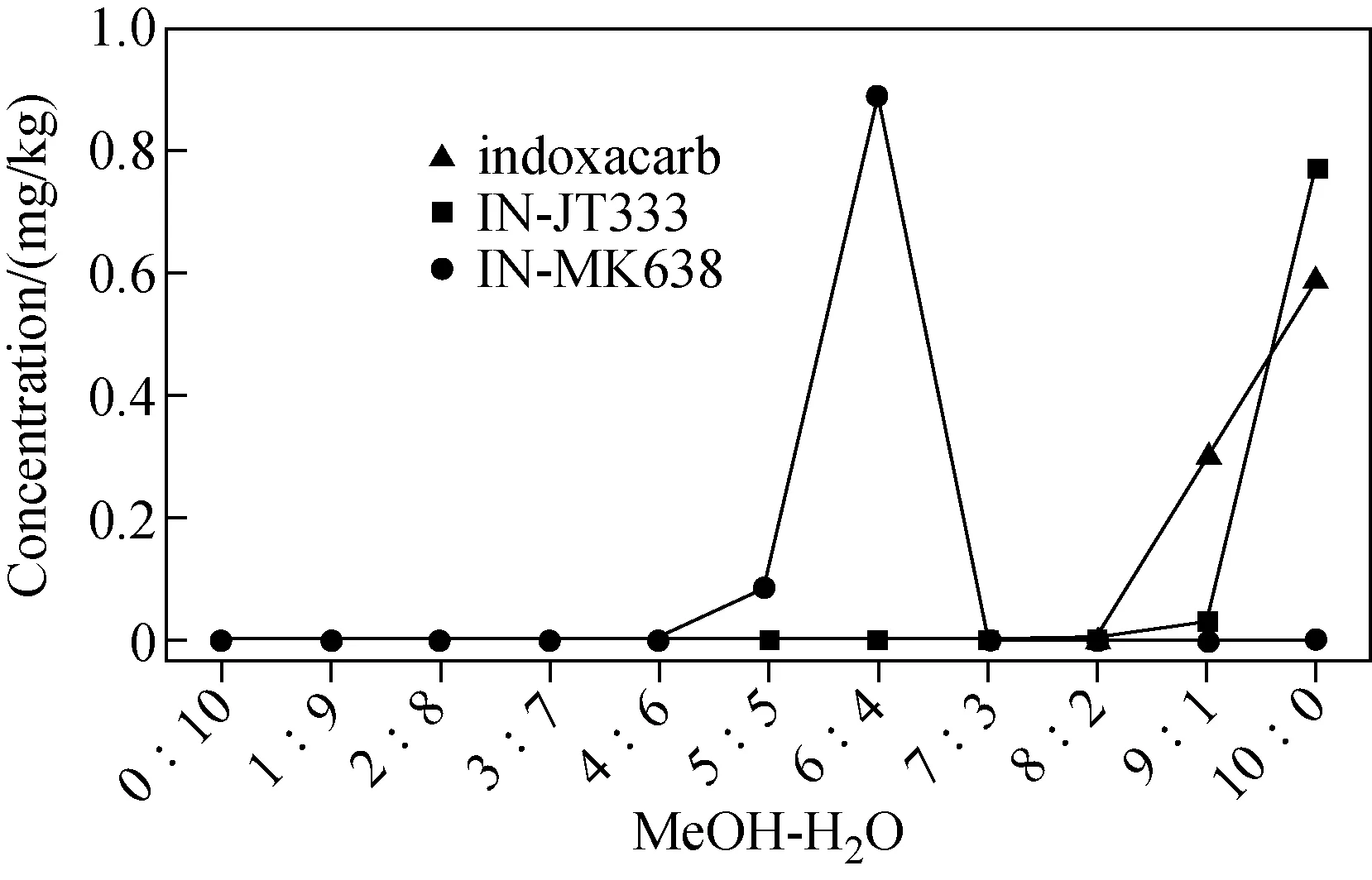
图2 不同比例甲醇-水溶液洗脱下茚虫威及其两种降解产物在PRP柱上的流出曲线Fig.2 Elution curves of indoxacarb and its metabolites on PRP column by MeOH-H2O with different ratios

图3 不同比例甲醇-水溶液洗脱PRP柱上茶汤样品的净化效果Fig.3 Purification effect of tea infusion sample on PRP column by MeOH-H2O with different ratios
2.2 色谱-质谱条件的优化
在前期研究[23]中,虽然采用Lux®3 μm Cellulose-1柱实现了对茚虫威对映体的拆分,也明确了乙腈拆分效果不如甲醇。但是本研究中发现Lux®3 μm Cellulose-1柱很难实现对IN-JT333对映体的拆分。通过重新对比其他手性柱的效果,发现Lux®3 μm Cellulose-2既能实现对茚虫威对映体的拆分,又能实现对IN-JT333对映体的拆分和对IN-JU873对映体的基本并肩峰分离;两种色谱柱上茚虫威的2个对映体流出顺序不同,在Lux®3 μm Cellulose-1柱上茚虫威-R体比茚虫威-S体先洗脱出来,而在Lux®3 μm Cellulose-2柱上则相反,这主要是由两种色谱柱填料中苯环取代基及位置不同而造成的选择性分离差别。最终选择Lux®3 μm Cellulose-2柱实现对茚虫威对映体及其降解产物的分离。
采用UPLC-ESI+-MS/MS对茚虫威及其7种降解产物进行分析,优化锥孔电压和喷雾电压,分别得到准分子离子峰[M+H]+,对准分子离子峰进行二级质谱子离子扫描,得到碎片离子信息,优化碰撞裂解能量,20 eV碰撞能量下茚虫威及其7种降解产物的ESI+-MS/MS质谱图示于图4,最终优化后二级质谱条件列于表1。
2.3 标准曲线、灵敏度和基质效应
在2.1和2.2节优化的实验条件下,测定茚虫威对映体及降解产物在0.01、0.025、0.05、0.10、0.25、0.50、1.0、2.5、5.0、10 mg/L(对映单体浓度为一半)浓度范围的进样溶剂、茶鲜叶、红茶、绿茶、红茶茶汤和绿茶茶汤基质标准溶液,得到线性方程和相关系数,列于表3。结果表明,在上述各种基质中,茚虫威对映体及其降解产物线性关系良好,相关系数(R2)均在0.999 1以上,能够满足检测要求。
茶叶等样品经过净化后,还会存在一定的基质减弱效应,因此需要采用基质标准外标法定量分析。UPLC-MS/MS测定茚虫威对映体及其降解产物的典型色谱图示于图5。
2.4 回收率和精密度
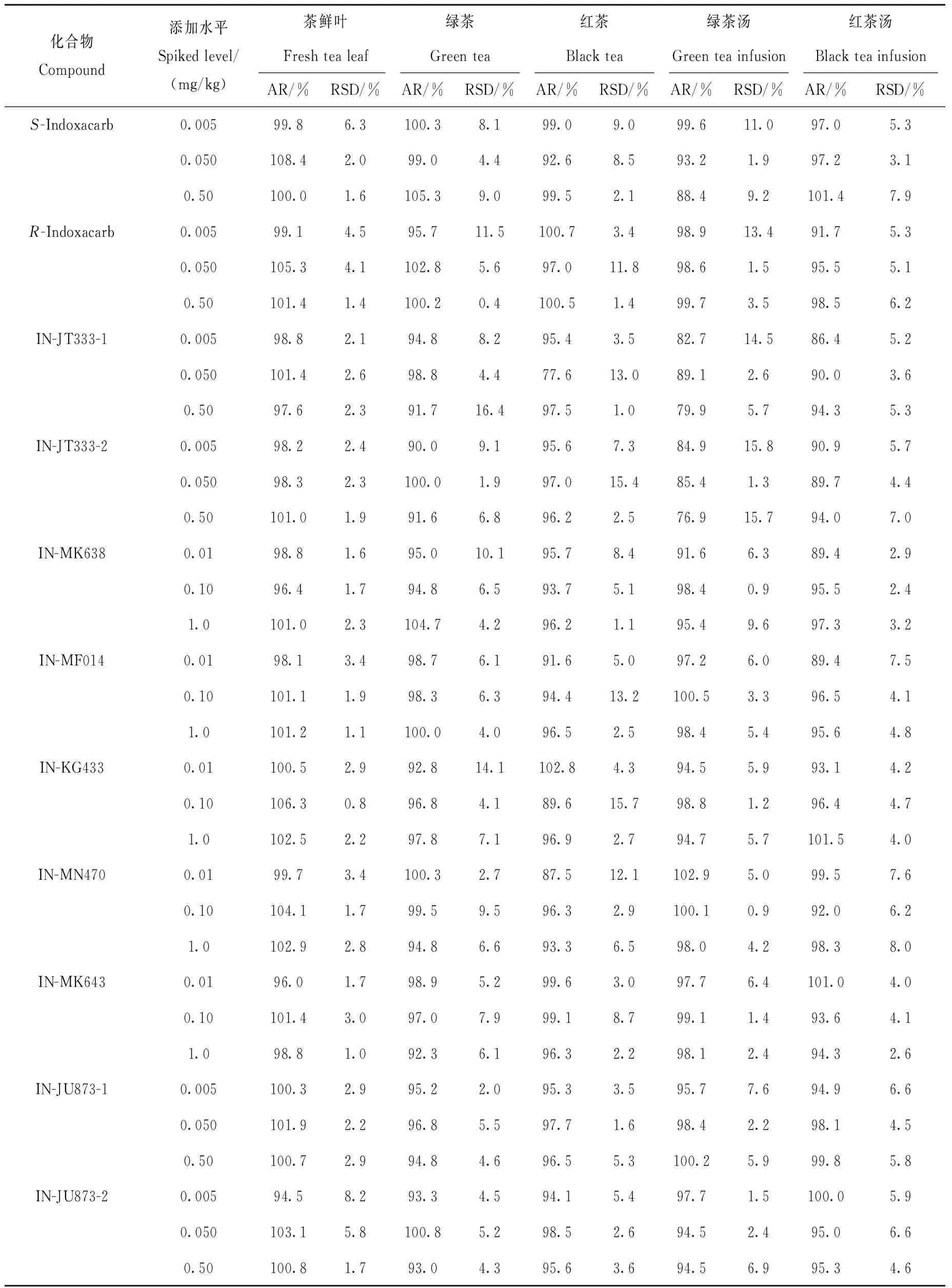
按照1.6节方法对茶鲜叶、绿茶、红茶、绿茶茶汤、红茶茶汤中茚虫威对映体及降解产物进行低、中、高浓度水平下6次平行添加回收率实验,结果列于表4。结果表明,在0.005、0.05、0.50 mg/kg(茶汤中为0.5、5、50 μg/L)添加浓度下,茚虫威-S体和茚虫威-R体、IN-JT333-1和IN-JT333-2、IN-JU873-1和IN-JU873-2在茶鲜叶中的平均添加回收率为94.5%~108.4%,RSD为1.6%~8.2%;在绿茶中的平均添加回收率为90.0%~105.3%,RSD为0.4%~16.4%;在红茶中的平均添加回收率为77.6%~101.3%,RSD为1.0%~15.4%;在绿茶汤中平均添加回收率为76.9%~100.2%,RSD为1.3%~15.8%;在红茶汤中平均添加回收率为86.4%~101.4%,RSD为3.1%~7.9%。
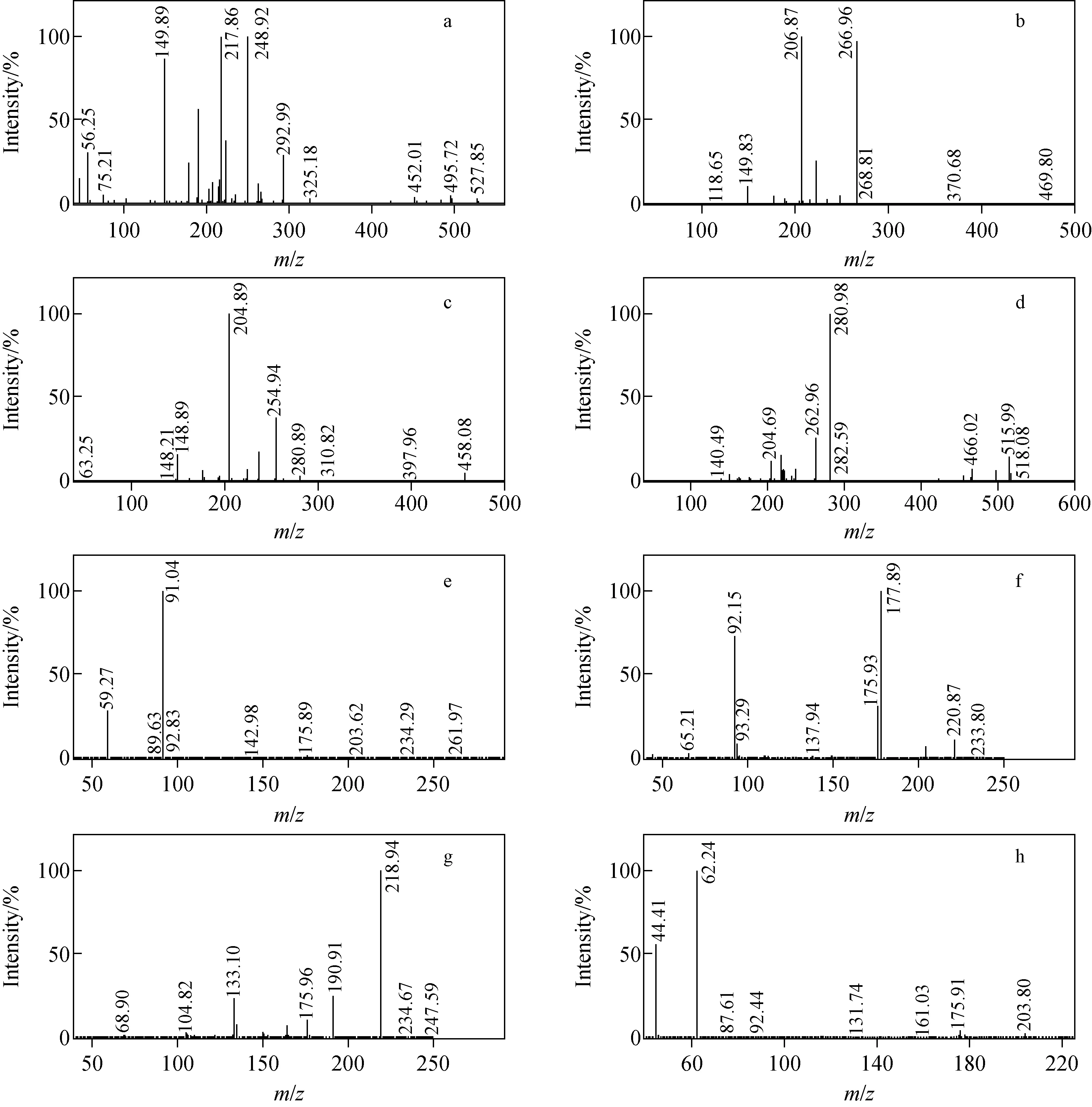
注:a.Indoxacarb;b.IN-JT333;c.IN-JU873;d.IN-KG433;e.IN-MF014;f.IN-MK638;g.IN-MK643;h.IN-MN470图4 20 eV下茚虫威及其降解产物母离子的UPLC-MS/MS二级质谱图Fig.4 MS/MS spectra for parent ions of indoxacarb and its metabolites at 20 eV
在0.01、0.10、1.0 mg/kg(茶汤中为1、10、100 μg/L)添加浓度下,IN-MK638、IN-KG433、IN-MN470、IN-MK643和IN-MF014在茶鲜叶中的平均添加回收率为96.0%~106.3%,RSD为0.8%~3.4%;在绿茶中的平均添加回收率为92.3%~104.7%,RSD为2.7%~9.5%;在红茶中的平均添加回收率为89.6%~99.1%,RSD为1.1%~15.7%;在绿茶汤中平均添加回收率为91.6%~102.9%,RSD为0.9%~9.6%;在红茶汤中平均添加回收率为89.4%~101.5%,RSD为2.4%~8.0%。
2.5 方法检出限与定量限
按照最低添加浓度水平下的响应,计算信噪比S/N=3时,所得各化合物在茶鲜叶、红茶、绿茶和茶汤等不同基质中的检出限均小于0.002 mg/kg,以满足回收率和相对标准偏差要求下的最低添加浓度水平来定义方法定量限(LOQ)。茚虫威-S体和茚虫威-R体、IN-JT333-1和IN-JT333-2、IN-JU873-1和IN-JU873-2在鲜叶、绿茶和红茶中LOQ为0.005 mg/kg,在绿茶汤和红茶汤中LOQ为0.5 μg/L;IN-MK638、IN-MF014、IN-KG433、IN-MN470和IN-MK643在茶鲜叶、绿茶和红茶中LOQ为0.01 mg/kg,在绿茶汤和红茶汤中LOQ为1 μg/L。
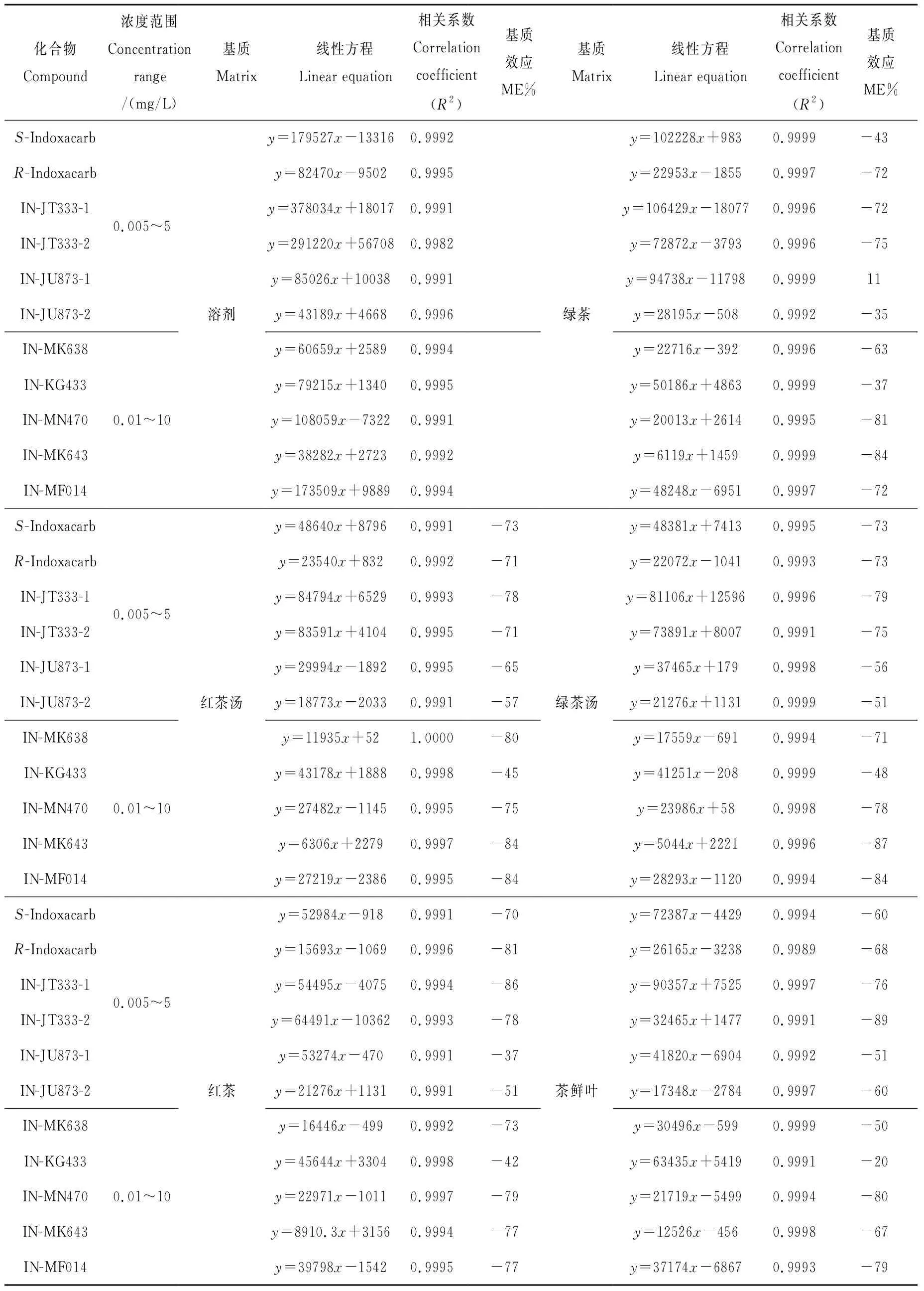
表3 UPLC-MS/MS测定不同基质溶液中茚虫威对映体及其降解产物的线性方程、相关系数和基质效应Table 3 Linear regression equations, correlation coefficients (R2) and matrix effects (MEs) of indoxacarb enantiomers and its metabolites in different matrix by UPLC-MS/MS
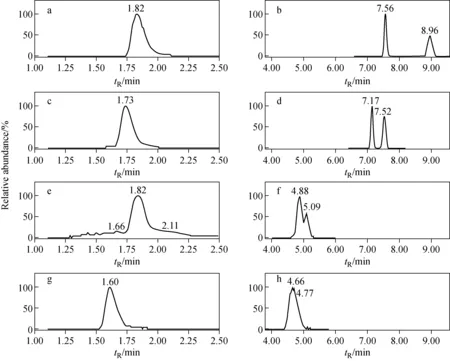
注:a.IN-MF014;b.IN-JT333;c.IN-MK638;d.Indoxacarb;e.IN-MK643;f.IN-JU873;g.IN-MN470;h.IN-KG433图5 茚虫威对映体及其降解产物(0.10 mg/L)的UPLC-MS/MS色谱图Fig.5 Chromatograms of indoxacarb and its metabolites (0.10 mg/L) by UPLC-MS/MS
2.6 实际样品测定
利用本方法对茶园茶叶喷施100 g/hm2剂量茚虫威S富集体(3S+1R)后,采集喷药后2 h、2天、5天、7天、10天和14天的茶鲜叶样品,冷冻干燥后粉碎,进行检测,通过保留时间和质谱离子对比对定性,在样品中检出有茚虫威-S体、茚虫威-R体以及IN-JT333-1、IN-MF014和 IN-KG433 3种降解产物,用基质外标法定量得出样品中上述化合物残留量分别为0.027~2.92 mg/kg、0.007~0.89 mg/kg、0.009~0.050 mg/kg、 本研究通过优化提取净化条件,采用乙腈-水溶液提取鲜叶、红茶和绿茶样品,C18与GCB分散固相萃取净化,浓缩后用甲醇-水溶液定容;对茶汤采用PRP柱富集净化淋洗,甲醇洗脱接收,浓缩近干后甲醇-水溶液定容,使用Lux®3 μm Cellulose-2柱对茚虫威对映体及7种降解产物进行色谱分离,UPLC-MS/MS基质外标法定量,检测茶鲜叶、绿茶、红茶、绿茶汤和红茶汤等基质中茚虫威对映体及其7种降解产物的残留。不同基质中所有化合物的标准曲线线性相关系数均在0.999以上,平均添加回收率为76.9%~108.4%,相对标准偏差小于16.4%,方法的定量限不高于0.01 mg/kg(1 μg/L)。本方法能够满足残留分析要求,为茶鲜叶、茶叶和茶汤中茚虫威对映体及其降解产物的研究检测提供方法参考。 表4 UPLC-MS/MS测定不同样品中茚虫威对映体及其降解产物的添加回收率和相对标准偏差Table 4 Average recoveries and relative standard deviations (RSDs) of indoxacarb and its metabolites in different samples by UPLC-MS/MS3 结论