基于铱(III)催化的吲哚螺β-内酰胺的构建
2021-03-31何旺白红进
何旺,白红进
(塔里木大学生命科学学院/新疆兵团南疆化工资源利用工程实验室,新疆 阿拉尔 843300)
含氮有机分子化合物,由于其广泛的生物学性质和药理学活性而受到越来越多合成和药物化学研究者的关注,其中,β−内酰胺更是因为其显著的生物活性而成为研究热点[1]。早在1940年,青霉素就作为第一个β−内酰胺类抗生素投入使用,目前广泛使用的抗生素均有β−内酰胺环结构,主要分为四类:青霉素类[2]、头孢菌素类[3]、碳青霉烯类[4]和单环β−内酰胺类[5]。可以说,在一定程度上,β−内酰胺类抗生素影响着人类社会的发展。近年来,抗生素的耐药性问题日趋严重,设计合成带有β−内酰胺结构的新型化合物势在必行。最新研究表明,β−内酰胺可以作为丝氨酸蛋白酶抑制剂[6]和酰基辅酶A胆固醇酰基转移酶(ACAT)的抑制剂[7],引起了许多课题组对β−内酰胺类化合物的合成兴趣[8−10]。事实上,自1907年开始,Staudinger首先合成出源自3−氨基丙酸的四元环内酰胺,之后 SKIKD J W 等[11]、BARBA V等[12]、KHASANON A B 等[13]、ZHANG R B 等[14]、ENDERS D等[15]和MIKHAIL S N等[16]课题组都对β−内酰胺类化合物进行了合成研究。本文以铱(III)催化吲哚噁唑酮类衍生物合成,经过氮−铱卡宾中间体来构建吲哚螺β−内酰胺,可以快速、简洁、高效的合成β−内酰胺,可为后续化学生物学研究提供物质基础。
1 实验部分
1.1 仪器与试剂
仪器:SOP型数显电子天平,赛多利丝科学仪器有限公司−北京;OSB−2100旋转蒸发器,上海爱朗仪器有限公司;SHB−Ⅲ循环水式真空泵,郑州长城科工贸易有限公司;85−2A型磁力搅拌器,上海司乐仪器有限公司;ZF−20D式暗箱型紫外分析仪,上海宝山顾村电光仪器厂;WS70−1型红外快速干燥箱,郑州市亚荣仪器有限公司;Bruker AMX−500型核磁共振仪。氘代试剂的溶剂残留标定为:CDCl3δ:7.26 ppm1H NMR,77.16 ppm13C NMR;d−acetone δ:2.05 ppm1H NMR,29.84 ppm13C NMR。
无水试剂的制备:无水乙醚和四氢呋喃以金属钠作为干燥剂,在氩气保护下蒸馏制取。快速柱层析在硅胶H或Silicycle P60硅胶上进行,石油醚(60~90℃),硅胶板 HSGF 254(烟台江友),紫外254显色或者磷钼酸的乙醇溶液加热显色。氘代试剂的溶剂残留标定为:CDCl3 δ:7.26 ppm 1H NMR,77.16 ppm 13C NMR;d−acetone δ:2.05 ppm 1H NMR,29.84 ppm 13C NMR。其它实验所用试剂均来源于商业途径,均为分析纯。
1.2 合成
产率的计算方法:主要产物的摩尔量/主要起始原料的摩尔量*100%;1的制备以吲哚乙酸3为主要起始原料,吲哚螺β−内酰胺2的制备以吲哚噁唑酮1为起始原料。
1.2.1 吲哚噁唑酮1的制备(如图1)
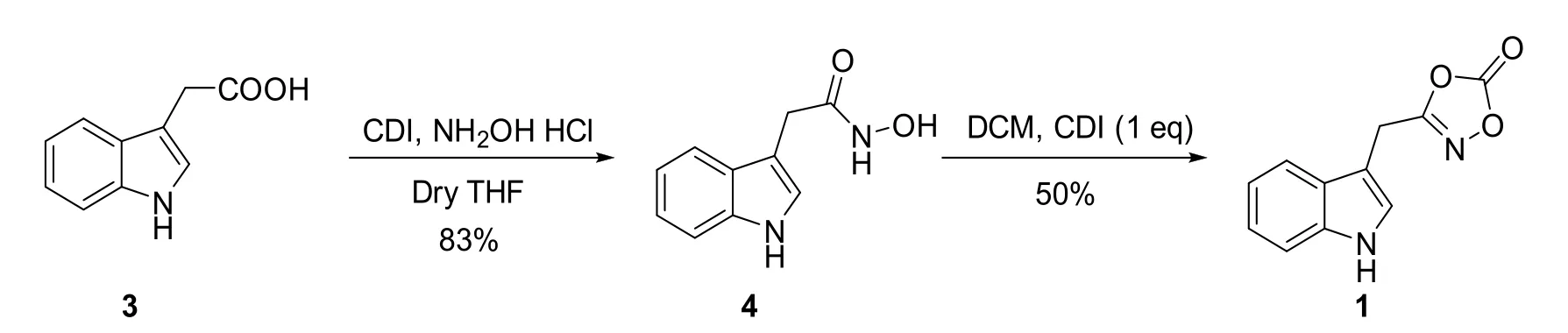
图1 吲哚噁唑酮1的制备
室温,在氩气保护下,将吲哚乙酸3(875 mg,5 mmol)溶于 THF(50 mL)中,接着将 CDI(970 mg,6 mmol)加入到上述反应体系中,搅拌半小时,然后将盐酸羟胺4(525 mg,7.5 mmol)加入反应体系中。反应24 h后检测反应,底物反应完毕,加入水(20 mL)淬灭反应,充分搅拌,分离有机相,用乙酸乙酯(3次×20 mL)萃取水相,合并有机相,用饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤,浓缩,柱层析纯化得到白色固体(788 mg,产率为83%,Rf=0.3,乙酸乙酯/甲醇=20:1)。该产物直接用于下一步反应,在氩气保护下,肟酸(570 mg,3 mmol)悬浮于DCM(25 mL)中,室温搅拌5 min后,将CDI(485 mg,3 mmol)加入反应液后室温继续反应1 h。反应结束后,浓缩,柱层析纯化得到无色油状物(320 mg,产率为50%,Rf=0.5,石油醚/乙酸乙酯 =10:1)。1H NMR(500 MHz,CDCl3)δ:8.22(s,1H),7.62(d,J=8.0 Hz,1H),7.42(d,J=8.0 Hz,1H),7.29~7.26(m,1H),7.22~7.19(m,2H),4.11(s,2H).13C NMR(125 MHz,CDCl3)δ:165.6,154.3,136.3,126.5,123.8,123.1,120.6,118.4,111.7,104.9,21.8.HRMS(ESI−TOF)m/z:[M+H]+Calcd.for C11H8N2O3217.0607,found:217.0608.
1.2.2 吲哚螺β−内酰胺2的制备(如图2)

图2 β-内酰胺的合成
在氩气保护下,将铱催化剂(3 mg,0.005 mmol)与NaBARF4(四(3,5−二(三氟甲基)苯基)硼酸钠(6.2 mg,0.007 mmol)溶于0.5 mL HFIP中,室温下搅拌反应10 min,随后将溶于0.5 mL HFIP中的化合物1(21.6 mg,0.100 mmol)加入到上述反应液中,随后将吲哚(23.4 mg,0.200 mmol)加入上述反应体系中搅拌反应36 h,监测反应结束后,将溶剂旋蒸至干,二氯甲烷溶解,柱层析纯化得到白色固体2。
吲哚螺β−内酰胺 2,(11.3 mg,产率为39%,Rf=0.4,石油醚/乙酸乙酯 =1:2)。1H NMR(500 MHz,CD⁃Cl3)δ:10.38(s,1H),8.17(d,J=8.5 Hz,1H),7.42 ~ 7.39(m,3H),7.36~7.33(m,1H),7.18(d,J=8.0 Hz,1H),7.10~7.06(m,2H),6.93(s,1H),6.89(t,J=7.5 Hz,1H),5.85(s,1H),5.40(s,1H),3.53(dd,J=15,1.5 Hz,3H),3.16(dd,J=15,1.5 Hz,1H).13C NMR(125 MHz,CDCl3)δ:166.4,156.3,145.1,138.2,132.4,130.4,126.6,125.0,123.3,123.2,122.7,120.3,120.2,116.1,112.6,65.4,63.2,55.9.HRMS(ESI−TOF)m/z:[M+H]+Calcd.for C18H16N3O:290.1286,found:290.1286.
2 结果与讨论
2.1 条件优化
本文以吲哚噁唑酮1为模板底物进行了条件的筛选(表1)。室温下,底物1在8−氨基喹啉衍生的铱催化剂下以NaBARF4为阴离子拮抗剂,以六氟异丙醇(HFIP)作为溶剂,吲哚作为亲核试剂,可以39%的产率得到吲哚螺β−内酰胺2(表1,处理1)。
铱催化剂的筛选:首先对具有不同氨基(7,5)保护基的催化剂进行了筛选,氨基由对甲苯磺酰基保护的铱催化剂未表现出较好的催化活性,内酰胺2的产率偏低(表1,处理2)。甲氧羰基(Meoc)保护氨基的铱催化剂可以35%的产率得到内酰胺产物2(表1,处理3)。将催化剂类型更换为8−羟基喹啉类型衍生的催化剂时,其催化效果仍未得到明显的提升(表1,处理4),故选定催化剂Ir1为铱催化剂来进行条件的优化。
溶剂的筛选:经过对各类溶剂(DCM,DCE,PhCF3,PhCl)进行了尝试,结果表明其反应效果均不如HFIP,故选择HFIP为最优溶剂进一步对反应条件进行探索(表1,处理5~8)。阴离子拮抗剂的筛选结果表明各类银盐在该反应中效果较差,推测是由于该类银盐酸性较强导致了底物噁唑酮的分解(表1,处理9)。进而对反应温度进行了初步尝试,结果表明0℃与50℃下,该反应的催化效果与产率都有所下降(表1,处理10~11)。
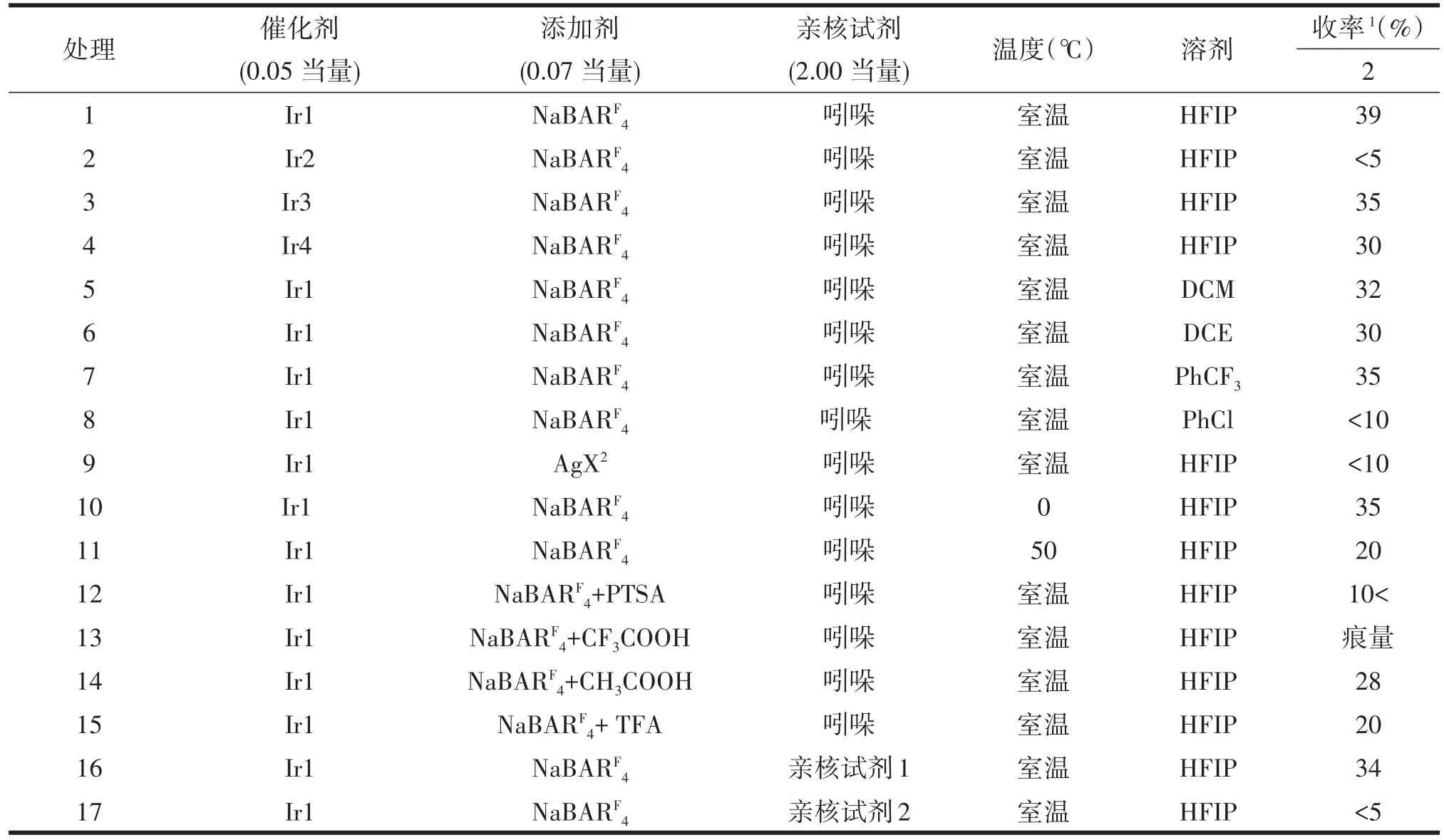
表1 条件筛选
添加剂的筛选:各类酸的加入除乙酸表现出相对较好的催化活性以外,其余酸(PTSA,CH3COOH,TFA)都未表现出优于最初条件的催化效果。最后,对亲核试剂进行了尝试,结果其反应性与选择性都较差。经过各类条件的筛选,得到了可一锅法构建吲哚螺β−内酰胺与吲哚酰胺的方法。
2.2 反应机理探究
将亲核试剂慢慢加入到反应体系内,结果发现吲哚酰胺2近乎为无法生成,由此推断其主要是环化时位阻较大且吲哚β−内酰胺的环张力偏大,不易形成。其推测机理如下:首先,噁唑酮与铱催化剂作用,释放出一分子CO2生成N卡宾中间体A,接着,吲哚中的氮原子电子反馈,吲哚中3−位碳作为亲核体对N卡宾进行亲核加成生成螺环亚胺中间体B,进而另一分子吲哚作为亲核试剂对亚胺进行亲核加成形成中间体C,进而质子解离铱催化剂生成螺环内酰胺2(图3)。
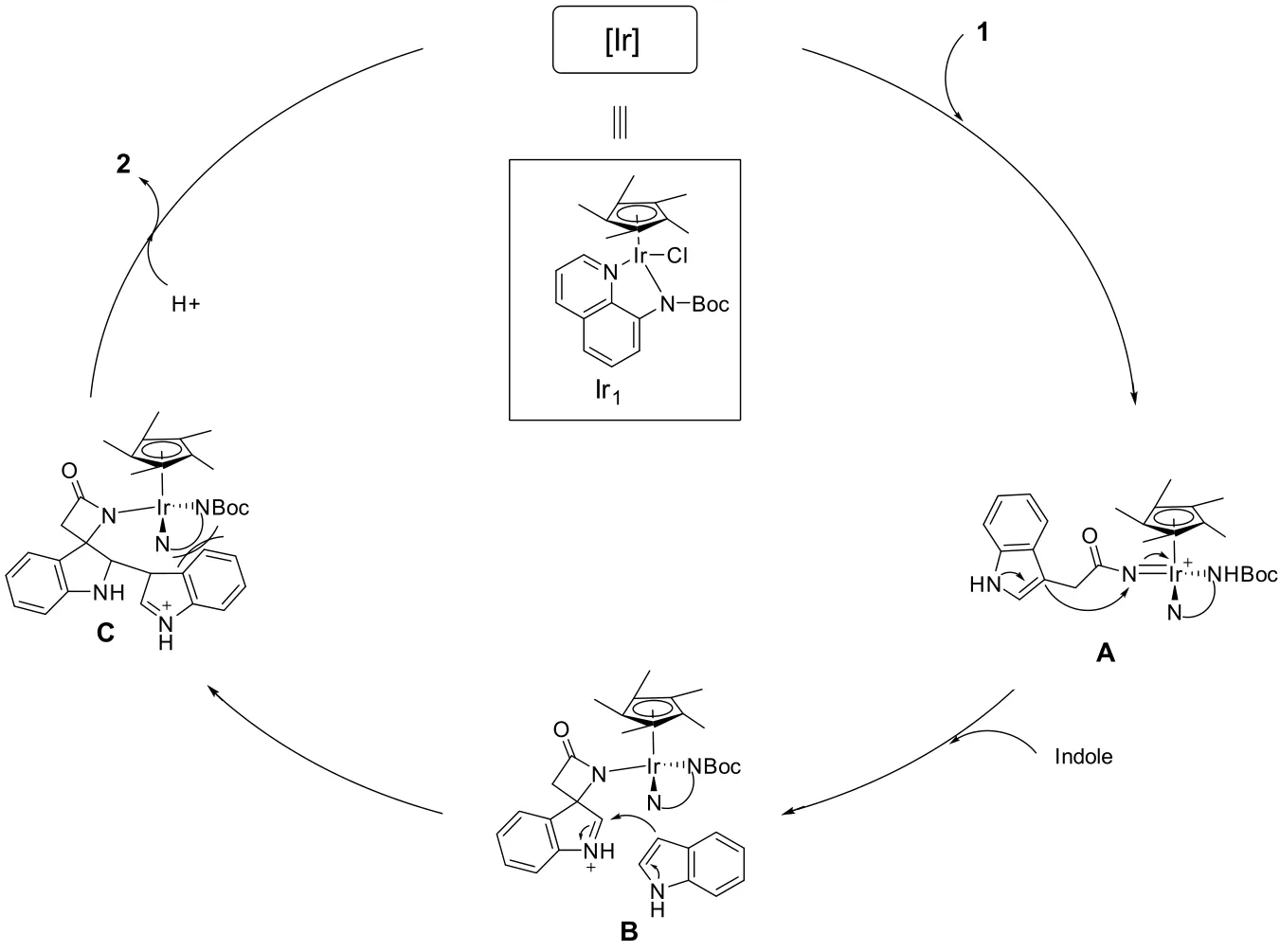
图3 机理推测
3 结论
目前,β−内酰胺的合成方法最经典的为Stauding⁃er β−内酰胺的合成以及近几年来开发的由金属催化的复杂分子内或分子间环化反应,其中包括分子内铜催化的α卤代烯胺环化[17],铑催化酰胺重氮化合环化[18]以及分子间钯催化腙与亚胺环化[19],锌催化亚胺与烯酮环化[20]。以上方法来构建β−内酰胺类化合物,其底物设计与合成以及反应体系较为复杂,且反应条件较为严苛。本课题组开发的铱催化噁唑酮进行分子内环化来构建此类内酰胺类化合物的方法,底物合成简单高效,且反应条件温和,易于操作,可一锅法合成复杂吲哚β−内酰胺类化合物,为后续β−内酰胺的合成提供一种新的思路及合成方向。
