以神经系统受累为主的Adams-Oliver 综合征2 型临床和基因分析
2021-03-29徐敏何燕郭虎
徐 敏 何 燕 郭 虎
南京医科大学附属儿童医院神经内科(江苏南京 210008)
Adams-Oliver综合征(Adams-Oliver syndrome,AOS)是一种罕见的多系统受累的遗传性疾病,发病率约0.44/100000,临床表现以先天性皮肤发育不良和肢体末端横向缺损为特征。此外,AOS还可伴有其他系统受累,如心血管系统、神经系统和眼部异常等。自1945年Adams和Oliver首次描述以来,至今已发现6 个致病基因:ARHGAP 31、DOCK 6、RBPJ、EOGT、NOTCH1和DLL4,分别导致AOS1~6型。本研究回顾分析1 例由DOCK 6基因变异导致的AOS 2 型患儿的临床资料。
1 临床资料
患儿,女,3岁1个月。因反复抽搐2年余,于2020年6月10日至南京医科大学附属儿童医院神经内科门诊就诊。患儿自幼发育落后,5个月会抬头,至今不能独坐,不会走路,与人眼神交流少,理解力差,只会说“妈妈”等简单叠词。外院曾予康复训练,无明显进步。患儿于6月龄时开始出现抽搐,首次发作以癫痫持续状态起病,抽搐表现为双眼上翻,口角流涎,四肢强直抖动,间断发作约1小时,发作间期意识不能恢复。首次发作后当地医院诊断癫痫。先后口服左乙拉西坦、丙戊酸钠抗癫痫治疗,抽搐不能完全控制,约1~2个月发作1次,发作形式与之前类似,每次持续约2分钟自行缓解,如遇感染发热时抽搐加重。患儿系G2P2,足月顺产,孕期及围生期无异常。父母体健,否认近亲结婚,姐姐11岁,体健。入院体格检查:神志清楚,不能听懂简单指令,眼神交流少,呼吸平,面色正常,头围46 cm(P3),头部皮肤、毛发未见缺损,全身皮肤未见色素脱失斑,双侧大腿外侧局部皮肤毛细血管扩张(图1),眼距稍宽,心、肺、腹查体未见异常,四肢肌张力明显增高,抬头尚稳,不能独坐,扶站时踮脚尖,四肢指(趾)末端未见缺损。实验室检查:血常规、血生化无异常;甲状旁腺素无异常;TORCH抗体阴性。头颅CT示两侧脑室旁多发钙化结节(图2),胼胝体发育不良,右侧颞叶低密度影。心脏彩超未见异常。头颅磁共振成像(MRI)示两侧脑室壁、室管膜下多发钙化,伴脑室旁片状脱髓鞘(图3),脑发育不良,双侧海马轻度萎缩,右侧颞枕叶异常信号。视频脑电图示右侧枕、后颞区多量中-高波幅棘慢波、多棘慢波发放,可波及对侧(图4)。眼底检查未见异常。视觉诱发电位未见异常。患儿于34月龄时Gesell发育量表评估示发育商(DQ)粗动作77,细动作76,应物能71,语言能52,应人能73。

图1 大腿外侧局部皮肤毛细血管扩张
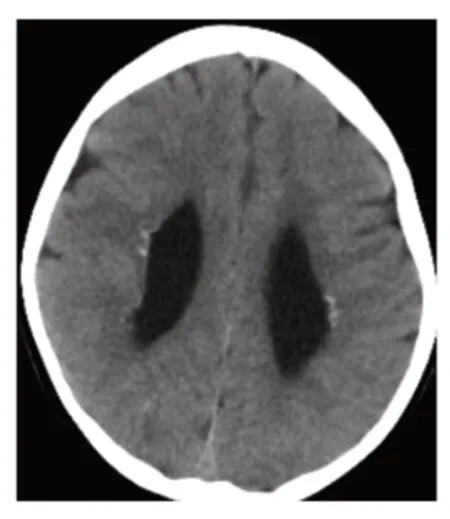
图2 患儿头颅CT 表现
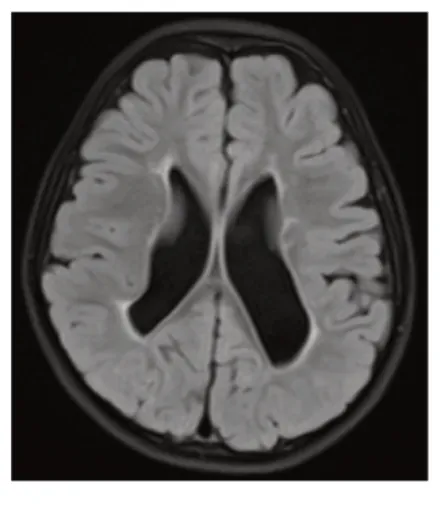
图3 患儿头颅MRI 表现
为进一步明确诊断,经南京医科大学附属儿童医院伦理委员会批准,患儿监护人知情同意,采集患儿及其父母外周血标本各2 mL,送至北京迈基诺医学检验所。采用德国Qiagen的QIAamp DNA Blood Mini Kit 试剂盒,提取基因组DNA。取患儿基因组DNA进行文库构建、全外显子捕获,使用Illumina公司 NovaSeq 6000系列测序仪进行高通量测序。使用BWA、GATK、Annovar等软件进行变异位点筛选,并采用Sanger测序法进行位点验证及家系验证。测序结果示患儿DOCK6基因c.3069_3069del和c.5220+1G>A,目前未见报道。两处变异均可能导致mRNA异常剪接(PVS1),该变异在gnomAD数据库中未收录(PM2),在变异的反式位置存在可能致病性变异(PM 3),DOCK 6基因相关疾病为AOS 2 型,与受检者临床表现相符(PP 4),参考ACMG 基因突变解读指南,2 处位点评级均定为致病性变异。Sanger测序结果显示,c.3069_3069del来自于父亲,c.5220+1G>A来自于母亲,父母均为杂合携带,患儿为复合杂合变异(图5),符合常染色体隐性遗传规律。
结合患儿临床症状及其他辅助检查,最终诊断为AOS2型。
2 讨论

图4 患儿脑电图表现
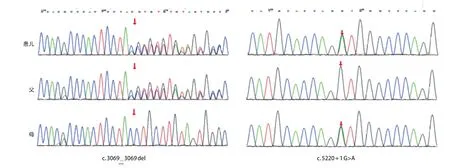
图5 患儿及父母基因测序结果
AOS分为1~6型,AOS最经典的表现为先天性皮肤发育不全和肢体末梢横向缺损,随着报道的增多,发现越来越多的临床表型,可累及全身多个系统,如心血管系统、眼部、消化系统、肾脏及神经系统等,出现各种先天性心脏病(室间隔缺损、肺动脉高压和圆锥动脉干畸形等)[1],视力障碍(内斜视,白内障,视网膜血管发育异常等),门静脉高压、食管静脉曲张,小肾、肾脏皮质血管异常等表现[2],神经系统受累最常见表现是癫痫发作。有研究显示,12 例AOS 患儿中,8例(66.7%)出现癫痫发作[3]。AOS亚型的临床表型与基因型有一定的相关性,如神经系统受累多见于DOCK 6和NOTCH 1基因变异,而在另外4 个基因则较少累及[4]。在各型AOS 患者中,AOS 2 型的临床异质性相对更为广泛[4],部分患者只有轻微的智力发育落后,而大部分患者有多系统受累;神经系统受累在AOS2型中尤为突出。2019年报道,19例AOS2患者中有16例出现神经系统异常[5]。AOS2型神经系统异常表现有小头畸形,不同程度的发育迟缓、抽动障碍、卒中、癫痫发作等[6]。已报道的大多数AOS 2 型患者有癫痫发作,甚至癫痫性脑病[7]。推测癫痫发作可能与脑发育异常有关,也不排除DOCK 6基因的神经元兴奋作用所致[3]。研究发现,即使同样的DOCK6基因变异,表型也会有差异。有报道在1 个AOS 2 型家系中,哥哥表现为轻度的趾甲发育不全、先天性心脏病、视力受损和认知障碍,而妹妹则表现为经典的先天性皮肤发育不全和肢体末梢横向缺损[8]。本例患儿临床表现更不典型,仅双侧大腿局部皮肤毛细血管扩张,缺乏AOS特征性肢体末端横向缺损,临床主要以神经系统受累为主,发育落后和癫痫发作为主要症状。本例患儿进一步扩大了AOS2型的表型谱。但患儿年龄小,日后是否会出现其他系统受累,还需长期随访观察。AOS 2 型的头颅影像学无明显特征性改变,大约3/4的病例出现头颅影像异常,包括大、小脑发育不全,皮层发育异常,脑室周围白质软化/钙化,胼胝体发育不良,脑室扩大,脑膨出和脑血管畸形(缺血、梗死)等[9-10]。本例患儿病初头颅CT示两侧脑室旁结节,与结节性硬化的头颅影像有一定的类似,最初于外院曾拟诊结节性硬化,进一步完善头颅MRI 检查示两侧脑室壁、室管膜下多发钙化,脑室旁片状脱髓鞘及脑发育不良、双侧海马萎缩,但是没有结节性硬化的其他症状,如皮肤色素脱失斑,且结节性硬化相关基因TSC1、TSC2未见异常,排除了结节性硬化。仔细分析患儿头颅MRI发现与既往文献报道AOS2的头颅MRI基本一致,但双侧海马萎缩尚未见文献报道。
自2011 年发现AOS 首个致病基因ARHGAP 31以来[11],至今已报道6 个明确的致病基因,但仍有40%~50%的AOS患者致病基因不明。在已发现的致病基因中,ARHGAP31、RBPJ、NOTCH1和DLL4为常染色体显性遗传,而DOCK6、EOGT为常染色体隐性遗传[12]。目前国内外已报道的导致AOS2的DOCK6基因变异30 余种,包括错义变异、无义变异、剪切位点变异、移码变异、大片段缺失[13]。本例患儿发现c.3069_3069del和c.5220+1G>A变异,目前国内外均未见报道。经ACMG评级均为致病性变异,扩大了DOCK6基因变异谱。AOS1~6型具体发病机制与不同的致病基因相关。AOS 2 型为DOCK 6基因变异所致,约占AOS 的13%~17%。DOCK 6基因变异导致Cdc42/Ral1信号通路失活,不能正确诱导肌动蛋白丝的聚合,导致胚胎形成过程中细胞不当凋亡和迁移,影响血管生成异常和细胞骨架重塑,导致肢芽发育异常,从而出现血管发育异常和手足横向缺损[14-15]。另外,脑血管在发育过程中受损是神经系统受累的发病机制之一[6]。
目前AOS的治疗无特殊,主要是针对各受累系统相应症状的对症处理,如器官畸形的外科干预,癫痫发作的抗癫痫治疗及发育落后的康复训练等。对于明确诊断的患儿,必要时需多学科联合长期管理。
综上,DOCK 6基因不同位点变异所致AOS 2 型患者,临床表型也不尽相同,本例患儿以神经系统受累为主,无肢端缺损表现。本研究扩大了AOS 2 型的DOCK 6基因变异谱。尽管目前缺乏有效治疗方法,早期明确诊断有助于家庭再生育的产前诊断和遗传咨询。
