沙门菌感染与Caspase-8介导的宿主细胞调控性死亡研究进展
2021-03-28原海波周丽婷吴超逸吴淑燕
原海波,周丽婷,吴超逸,吴淑燕
苏州大学医学部基础医学与生物科学学院,江苏 苏州 215123
沙门菌在自然环境中普遍存在,引起人类疾病的沙门菌通常与食品有关。基于其所致疾病的临床症状,沙门菌通常分为伤寒沙门菌和非伤寒沙门菌。伤寒沙门菌易引起肠热症,且其致病过程与非伤寒沙门菌不同,本文不做介绍,下文所述沙门菌均指非伤寒沙门菌。非伤寒沙门菌主要造成肠道感染(伴有腹泻、发热和腹部疼痛),一般具有自限性。但是当宿主免疫力下降时,沙门菌感染可导致败血症等严重后果。在沙门菌感染早期,宿主主要通过肠道上皮细胞和固有层巨噬细胞来限制沙门菌的定植和入侵。当宿主细胞被沙门菌感染后,细胞中的NLRC4(nucleotide-binding oligomerization domain, leucine rich repeat and CARD domain containing 4)炎性小体被激活,进而激活Caspase-1介导的消皮素D(gasdermin D,GSDMD)切割,触发细胞焦亡程序,诱导炎症反应[1-2],其他免疫细胞通过趋化作用到达炎症部位,可以吞噬清除沙门菌。研究发现,当经典细胞焦亡通路被抑制(即Caspase-1或GSDMD被抑制)时,宿主细胞可以通过强烈激活Caspase-8来诱导细胞死亡和炎症反应,从而限制沙门菌感染[3-4]。
细胞死亡的方式越来越多样化,新的观点认为程序性细胞死亡(programmed cell death, PCD)是一种特殊形式的调控性细胞死亡(regulated cell death, RCD),通常是生理性死亡,是维持组织内稳态所必需的。RCD是细胞死亡的一种形式,涉及特定的信号级联,因此可以通过基因或药物调控。RCD包括焦亡(pyroptosis)、凋亡(apoptosis)、坏死性凋亡(necroptosis)、铁死亡(ferroptosis)等多种细胞死亡方式[5]。沙门菌感染可导致细胞通过焦亡、凋亡等多种细胞死亡方式死亡。文献报道沙门菌毒力质粒编码的SpvC蛋白可以抑制丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)信号通路,进而抑制肠道炎症反应[6]。本课题组研究发现该效应与SpvC抑制宿主细胞焦亡有关,推测Caspase-8可能在沙门菌感染过程中被激活而发挥抗感染效应[7]。
本文主要介绍沙门菌感染期间NLRC4炎性小体被激活后发挥重要的抗感染免疫功能,Caspase-8参与NLRC4炎性小体的组装,可参与调节炎症因子的释放,同时通过调节宿主肠道上皮细胞和巨噬细胞RCD参与抗感染免疫。
1 沙门菌感染导致炎性小体激活和Caspase活化
宿主通过模式识别受体(pattern recognition receptor,PRR)来识别病原体相关分子模式(pathogen-associated molecular pattern,PAMP)。NOD样受体(nucleotide-binding and oligomerization domain-like receptor,NLR)是位于胞质内非常重要的一种PRR,它通过感知细胞内外源性病原微生物产物或内源性危险信号,启动包括炎性小体在内的多种蛋白复合体组装,引发抗感染免疫。NLR被激活后通常导致炎性小体形成,如NLRC4炎性小体、NLRP3炎性小体,它们可以通过其配体蛋白即凋亡相关斑点样蛋白(apoptosis-associated speck-like protein,ASC)募集Caspase-1和Caspase-8,Caspase-1对Caspase-8的活化具有抑制作用,通常情况下激活的Caspase-1切割GSDMD导致细胞焦亡和炎症反应;当Caspase-1被抑制时,Caspase-8可通过炎性小体通路活化启动凋亡;另一方面,Caspase-8激活的Caspase-3可切割另一种重要的焦亡执行蛋白gasdermin E,因此也可促进焦亡的发生。沙门菌感染宿主细胞后激活NLRC4、NLRP3等炎性小体,从而激活下游的Caspase-1和(或)Caspase-8,导致宿主细胞死亡和炎症反应以限制沙门菌感染(图1)。而沙门菌也通过各种方式延迟细胞死亡,抑制甚至利用宿主细胞炎症反应来促进自身生存。
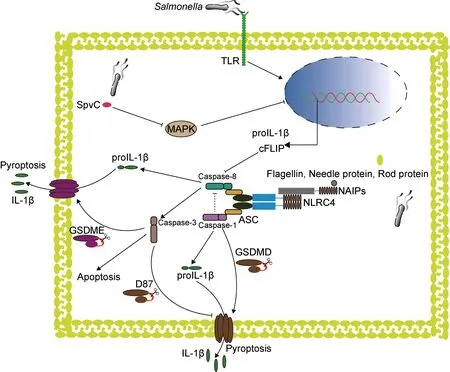
图1 沙门菌感染后Caspase-8被炎性小体激活
1.1 神经元凋亡抑制蛋白识别沙门菌并参与NLRC4炎性小体的激活
神经元凋亡抑制蛋白(neuronal apoptosis inhibitory protein,NAIP)是 NLR蛋白的一个亚家族,具有炎性小体受体的功能,可以直接识别来自病原菌的不同配体分子。小鼠可以编码7个 NAIP 家族的分子,其中4个分子(NAIP1、NAIP2、NAIP5和NAIP6)可以在通常使用的实验小鼠品系C57/BL6中表达。NAIP1和NAIP2能够分别识别革兰氏阴性细菌3型分泌系统(type 3 secretion system,T3SS)的针状蛋白和基座蛋白,NAIP5/6可以识别鞭毛蛋白[8-9]。与小鼠不同,人类编码单个功能性NAIP并能广泛检测多种革兰氏阴性细菌T3SS成分和鞭毛蛋白[10]。NAIP与其配体结合后,会与另一种NOD样受体NLRC4结合形成高分子量的环状低聚物,从而活化NLRC4。活化的NLRC4会发生自身多聚化并通过ASC募集Caspase-1形成炎性小体,从而诱导细胞因子分泌和细胞焦亡[11-12]。在沙门菌感染早期,与野生型小鼠相比,缺失NAIP或NLRC4的小鼠肠道上皮细胞中的沙门菌大量增加[4],并且在受沙门菌感染的结肠炎小鼠模型中,发现NLRC4的缺失导致小鼠体内菌量负载增加,生存率下降[13-14]。这些研究表明在沙门菌感染早期,NAIP/NLRC4炎性小体在宿主识别和限制沙门菌感染中发挥着重要作用。
1.2 沙门菌逃避和利用宿主免疫应答
沙门菌感染后炎性小体被激活,通常会导致炎症反应的发生,而炎症又是一把“双刃剑”,一方面,炎症刺激可清除病原体,另一方面,炎症的过度刺激可能导致机体功能紊乱,诱发新的病变。沙门菌有两个毒力岛,沙门菌毒力岛1(Salmonellapathogenicity island 1,SPI-1)和沙门菌毒力岛2(Salmonellapathogenicity island 2,SPI-2),分别编码两种T3SS,它们与沙门菌入侵宿主肠道上皮细胞、维持沙门菌在胞内存活[15-16]以及沙门菌持留菌的形成和耐药质粒的传播有关[17]。沙门菌通过以下方式影响宿主免疫。①避免被宿主识别。侵入细胞的沙门菌可以通过胞膜感受外界环境的变化,下调鞭毛蛋白的合成来逃避宿主NAIP的识别[18]。②抑制宿主细胞的炎症反应。沙门菌T3SS分泌的效应蛋白和毒力质粒编码的蛋白可以促进沙门菌的入侵,同时可以抑制细胞炎症反应进而维持沙门菌在胞内的存活[19-22]。比如沙门菌效应蛋白SopB不仅可以增强宿主细胞的胞吞作用,促进沙门菌入侵[23-25],还可以在B细胞中激活磷脂肌醇(PI3K)-蛋白激酶B(Akt)-Yes相关蛋白(YAP)通路,从而抑制NLRC4炎性小体的激活,逃避宿主免疫[26]。③利用宿主免疫促进自身生存。沙门菌感染引发急性肠道炎症,在此过程中产生的活性氧与内源性腔内硫化合物反应,所生成的连四硫酸盐能够被沙门菌利用,使沙门菌能够在与肠道正常菌群竞争中战胜依赖于厌氧发酵的其他微生物[27]。④沙门菌感染还能影响细胞内信号传递。例如:沙门菌效应蛋白SopB具有模拟细胞信号的能力,可以绕过宿主固有免疫受体直接激活Cdc42和PAK1,进而促进白细胞介素(interleukin,IL)-1β、肿瘤坏死因子(tumor necrosis factor,TNF)-α和IL-6的转录,抑制肠道中的固有菌群,促进沙门菌在肠道中复制[28];沙门菌效应蛋白SarA/SteE激活信号传导与转录激活因子3(signal transducer and activator of transcription 3,STAT3),促进沙门菌的胞内复制[29]。
沙门菌感染导致NLRC4炎性小体激活,Caspase-8和Caspase-1均参与了炎性小体的形成。研究表明,在沙门菌感染期间Caspase-8同样被激活[30],但其激活机制尚不明确。同时,研究发现Caspase-8参与调节炎症因子IL-1β的合成与成熟,并能够促进NLRP3炎性小体的激活[31-32]。
2 Caspase-8促进IL-1β前体合成和成熟以应对沙门菌感染
沙门菌感染早期,NLRC4炎性小体激活Caspase-1,活化后的Caspase-1可以加工IL-1β前体,产生成熟的IL-1β,从而限制沙门菌引起的肠道感染[33]。与Caspase-1相同, Caspase-8也可以被炎性小体激活,并加工IL-1β前体,产生成熟的IL-1β。当沙门菌感染小鼠骨髓起源巨噬细胞(bone marrow derived macrophages,BMDMs)时, NLRP3和NLRC4都被激活,并且与Caspase-1前体的加工和IL-1β的分泌密切相关;若两者均缺失,则会严重影响Caspase-1前体的加工和IL-1β的分泌[34]。这表明IL-1β前体的加工和分泌依赖于NLRP3和NLRC4的激活。研究发现,NLRC4被激活后可以招募NLRP3与ASC形成一个共同复合体,并招募Caspase-1和Caspase-8,在此复合体上,Caspase-1可以被激活并诱导细胞焦亡以及IL-1β前体的加工和分泌[35-37]。新的研究表明,NLRC4缺失导致小鼠在受到沙门菌感染时死亡率上升[13]。这可能是因为肠道黏膜固有层单核-吞噬细胞(intestinal mononuclear phagocytes,IMPs)不同于BMDMs,在脂多糖(lipopolysaccharide,LPS)刺激下不能提高NLRP3的表达,所以NLRC4在IMPs中发挥着重要功能。研究还发现IMPs可以通过NLRC4-IL-1R上调E-选择素等内皮黏附分子的表达,募集中性粒细胞来控制沙门菌在肠道中的感染[14]。Caspase-8与受体相互作用蛋白激酶3(receptor interacting protein kinase 3,RIPK3)、Fas相关的死亡结构域(Fas associated death domain,FADD)组装成的复合体可以促进IL-1β前体的成熟[38],Caspase-8也可以通过Fas信号介导以不依赖RIPK3和炎性小体的方式加工IL-1β前体[39]。促凋亡Bcl-2家族蛋白成员Bax和Bak诱导的细胞凋亡可触发Caspase-8介导的IL-1β前体的成熟[40],并且体外重组的Caspase-8可以在Caspase-1相同位点加工IL-1β前体,产生成熟的IL-1β[41]。经典细胞焦亡刺激物LPS+尼日利亚菌素刺激小鼠骨髓起源的树突细胞(dendritic cell,DC),可见Caspase-1被激活并介导IL-1β前体加工和分泌以及细胞焦亡,但在Caspase-1缺失时Caspase-8与NLRP3介导了IL-1β约2 h的延迟分泌[32],这可能因Caspase-8被激活后,通过其他信号通路诱导细胞膜孔形成,从而导致IL-1β的延迟分泌,但须进一步的实验证实。这些研究表明,Caspase-8被激活后同样具有加工和分泌成熟IL-1β的能力。所以,Caspase-8与IL-1β的合成和分泌密切相关,在一定条件下可以发挥部分Caspase-1功能,调节宿主免疫反应,限制沙门菌感染。但是值得注意的是,这种调节有一定的延迟性,并且相对于Caspase-1,Caspase-8促进IL-1β产生的能力较弱,而且Caspase-8下游是否发生其他生物学事件导致细胞死亡也须进一步研究。
3 Caspase-8通过参与宿主调控性细胞死亡限制沙门菌感染
虽然沙门菌通过各种方式在肠道内定植并侵入机体造成感染,但是宿主也可通过多种方式来限制其入侵和感染,而诱导受沙门菌感染的细胞死亡是一种有效的方式:一方面,死亡的细胞可以释放IL-1β、IL-18等炎症因子诱导炎症反应;另一方面,细胞死亡后破坏了胞内沙门菌的生存环境,更有利于沙门菌的清除。过去的几十年间,研究人员认为Caspase-8主要在细胞凋亡中发挥功能。细胞受到特定刺激后,Caspase-8通过自剪切激活,启动Caspase的级联反应,激活Caspase-3、6、7,最终导致细胞凋亡。随着研究的深入,Caspase-8在细胞焦亡和坏死性凋亡中的功能也不断被发现,其可被招募到不同的分子平台中发挥不同的生物学效应。研究表明,在肠道上皮细胞和巨噬细胞中,Caspase-8可被NLRC4炎性小体激活,诱导细胞死亡和IL-1β等炎症因子的释放,在限制和清除沙门菌感染中发挥重要作用[3,42-43]。作为肠黏膜屏障的一部分,肠道上皮细胞在沙门菌感染中发挥着不可替代的功能。肠道上皮细胞特异性基因敲除研究证实,Caspase-8分子或其酶活性的缺失都会促进炎性小体的形成,并导致严重的肠道炎症[44-45]。同时,Caspase-8可以通过维护上皮细胞屏障功能和限制沙门菌定植来阻止沙门菌诱导的肠道炎症[46]。与此一致的是,临床病例研究表明Caspase-8的突变可能是炎症性肠病发生的原因之一,并且Caspase-8突变的患者更易受到肠道病原体如沙门菌的侵袭[47]。上述研究表明,Caspase-8调控细胞死亡并诱导炎症反应的能力与沙门菌感染密切相关。
3.1 Caspase-8参与肠道上皮细胞调控性细胞死亡以应对沙门菌感染
在肠道组织中,与摄取沙门菌的DC相比,肠道上皮细胞表达更多的NAIP和NLRC4,这为肠道上皮细胞快速识别、清除沙门菌提供了功能基础[48]。肠道上皮细胞的NLRC4炎性小体被沙门菌激活后,促进类花生酸和炎症因子IL-18释放,类花生酸的释放可以诱导肠道炎症,一方面促进细胞内液丢失并驱动受感染的细胞脱落排入肠腔,另一方面导致腹泻以限制沙门菌在体内的数量[4,49]。研究表明,受感染的肠道上皮细胞死亡脱落与其周边细胞的肌动蛋白重排密切相关,并且肠道上皮细胞的死亡不完全依赖于Caspase-1/GSDMD介导的细胞焦亡,这是因为敲除Caspase-1/GSDMD虽然阻止细胞焦亡的发生,但是肠道病理性变化和肠道上皮细胞死亡脱落并未受限。进一步研究发现,通过ASC被募集到NLRC4炎性小体上的Caspase-8在缺失Caspase-1/GSDMD时可以被激活,导致受沙门菌感染的肠道上皮细胞死亡、脱落[3]。与此一致,另一研究表明当沙门菌感染缺失Caspase-1/11的肠道上皮细胞类器官时,Caspase-8被NLRC4炎性小体激活并诱导细胞凋亡,同时发现Caspase-1可以抑制Caspase-8的激活[50]。所以在肠道上皮细胞中,受到沙门菌感染的肠道上皮细胞快速脱落依赖于NLRC4炎性小体的功能,并且在Caspase-1介导的细胞焦亡通路受阻时,NLRC4炎性小体可以通过激活Caspase-8诱导的细胞凋亡来限制沙门菌的入侵。这可能为治疗NLRC4炎性小体突变导致的自身免疫性疾病提供了新的方法,即抑制Caspase-1后,细胞焦亡被转换为Caspase-8介导的细胞凋亡,从而减轻炎症反应[51]。但这也带来新的问题,已有研究表明Caspase-8的激活可能导致肠道上皮细胞发生其他类型的RCD而不只是细胞凋亡[3]。有研究表明,虽然沙门菌感染期间Caspase-8对细胞死亡影响有限,但是在小鼠肠道上皮细胞特异性敲除Caspase-8后,与野生型小鼠相比,沙门菌在肠道定植更多,并引发更严重的肠道炎症,导致宿主死亡[46],提示Caspase-8在某些条件下可通过目前尚不明确的方式限制病原菌入侵,维持肠道稳态。
3.2 Caspase-8参与巨噬细胞调控性细胞死亡以应对沙门菌感染
在BMDMs中,Caspase-1和Caspase-8被ASC募集,它们与ASC的结合是非竞争关系[35]。 Caspase-1酶活性缺失后, Caspase-8在NLRC4炎性小体上被激活并导致细胞凋亡,同时还发现细胞膜损伤,细胞裂解死亡,这可能是Caspase-3被Caspase-8激活后切割另一Gasdermin 家族成员GSDME所导致的细胞焦亡[52-53];但研究又发现,敲除GSDME后并不影响Caspase-8被激活后所诱导的细胞裂解死亡[30]。与此一致的是,NLRP3炎性小体也可以通过ASC募集Caspase-8和 Caspase-1,在缺失GSDMD且Caspase-1酶活性突变的骨髓起源的DC中,Caspase-8被强烈激活并介导IL-1β前体加工和细胞焦亡[43]。近期一项研究发现,小鼠BMDMs和RAW264.7细胞系在NLRP3被抑制的情况下,由ATP诱导的细胞焦亡会被阻断,此时Caspase-8被激活,从而导致细胞凋亡及因 Caspase-3 切割GSDME所诱导的细胞焦亡[54]。同时这些研究也发现,除GSDME介导的细胞焦亡外,还发生了其他形式的RCD。Caspase-8介导的细胞凋亡作为一种“沉默性”细胞死亡方式,不会引起炎症反应,从而推测可为沙门菌的扩散提供便利。过度的炎症反应会给宿主造成不良后果,因此机体可能通过调节Caspase-8这一关键分子,平衡不同类型的RCD,利用适度的炎症反应清除病原菌,维持机体的稳态。
与沙门菌相似,嗜肺军团菌(Legionellapneumophila)感染小鼠巨噬细胞时,其鞭毛蛋白进入胞质激活NLRC4炎性小体,活化的炎性小体同样可以募集Caspase-1和Caspase-8,但只有在Caspase-1或GSDMD被抑制时,Caspase-8才能被激活,并导致膜孔形成和细胞裂解死亡[55]。而小肠结肠炎耶尔森菌(Yersiniaenterocolitica)感染与沙门菌不同,它所造成的巨噬细胞死亡、Caspase-1激活和IL-1β释放主要取决于Caspase-8的激活,并且Caspase-8的激活与炎性小体无关[56-57]。这些研究表明宿主在应对不同病原体感染时,巨噬细胞通过不同方式激活Caspase-8会产生不同的生物学效应,研究这种差异性的产生机制对深入了解宿主免疫功能具有重要意义。最近研究发现,Caspase-8活性受到 Thr265磷酸化的调控[58],但是Caspase-8在炎性小体中的激活与调控机制仍不明确,其激活后发生的生物学事件对于细胞死亡方式的调控有待更进一步的研究。
4 总结与展望
Caspase-8在程序性细胞死亡中发挥着重要的调节功能,在沙门菌感染的细胞中,当细胞焦亡通路受到抑制时,Caspase-8会被激活以限制沙门菌复制,从而保障宿主清除沙门菌。本课题组研究发现,沙门菌质粒编码的效应蛋白SpvB在感染早期抑制nlrp3转录,但尚不清楚这种抑制机制是否与Caspase-8有关[31,59],同时发现沙门菌效应蛋白SpvB可以延迟细胞焦亡,而且沙门菌毒力质粒编码的SpvC效应蛋白可以通过抑制MAPK通路,进而抑制细胞焦亡[7]。本综述中探讨了沙门菌感染期间抑制细胞焦亡通路后可以导致Caspase-8激活,推测沙门菌毒力质粒编码的效应蛋白SpvB和SpvC延迟细胞焦亡的功能可能与Caspase-8相关,进一步研究其相互关系对于厘清沙门菌致病机制具有重要意义。
Caspase-8的活性也受到多种因素的影响[60],最近一项研究表明,抗凋亡蛋白(cellular FLICE-like inhibitory protein,cFLIP)可以抑制Caspase-8的活性进而调节细胞死亡和炎症反应[61]。FADD作为Caspase-8的适配蛋白,通过调控Caspase-8介导的炎症反应维持肠道细胞的稳态[62-63]。同样值得关注的是,Caspase-8在炎性小体上被活化后的下游事件。Caspase-3被Caspase-8激活后不仅可以诱导细胞凋亡,也可以导致其他生物效应。例如:细胞焦亡执行蛋白GSDMD可以被Caspase-3在D87处裂解,导致其在质膜上成孔的活性丧失[64];被激活的Caspase-3可以反馈激活上游的Caspase-8,进而放大凋亡事件[65];Caspase-3还可以切割DFNA5(GDMDE)诱导细胞焦亡[52-54]和内源性细胞凋亡[66]。而Caspase-8也可以直接切割GSDMD导致细胞焦亡[56-57]。所以,Caspase-8下游底物的激活导致细胞走向不同的命运,这也说明不同RCD之间是相互关联的,细胞最终的死亡方式可能取决于Caspases裂解的底物。未来的研究可能使用更具特异性的抑制剂,促进或抑制Caspase-8的活性,从而促进细胞死亡或抑制炎症,例如Caspase-8激活后促进细胞死亡可以作为肿瘤的治疗靶标[67]。通过抑制Caspase-8活性可以减轻蛛网膜下隙出血所导致的脑损伤和角膜移植后的排斥反应[68-69],这对进一步开发肿瘤和炎症性疾病治疗的新药物和方法具有重要意义。
