先天性肾病综合征1例报告及文献复习
2021-03-25陈丽颖李志鑫刘振球
陈丽颖 李志鑫 饶 睿 刘振球
四川省乐山市人民医院儿科 614000
先天性肾病综合征(CNS)通常指出生3个月内发病,临床表现符合肾病综合征(NS,大量蛋白尿、低白蛋白血症、严重水肿和高胆固醇血症)。现将收治的1例CNS患儿的临床表现、实验室检查、治疗经过、基因分析等进行总结,并复习相关文献,提高对该病的认识。
1 临床资料
1.1 一般资料 患儿女,日龄13d。因“双下肢浮肿、发现蛋白尿5+d”入院。患儿系第5胎,第2产,孕37+2周,单胎,因母亲妊娠合并急性胰腺炎行剖宫产,Apgar评分不详,产重3 320g,羊水Ⅲ度粪染,量30ml,脐带、胎盘无异常,否认宫内窘迫,无胎膜早破,无窒息史。生后人工喂养,体重无明显增长。家族史:患儿父亲35岁,母亲37岁,母亲有胃炎、胰腺炎、高血脂病史,父亲身体健康,非近亲结婚,有一姐,8岁,脑瘫。家人无类似病史,家族中无肾脏病患者。
1.2 检查 入院查体:体温36.5℃,脉搏132次/min,呼吸47次/min,血压95/53mmHg(1mmHg=0.133kPa),体重3.38kg,身长52cm。外貌成熟儿,反应欠佳,面色、肤色苍白,双眼睑轻度水肿,双肺听诊呼吸音稍粗,未闻及干湿啰音。心率132次/min,心音欠有力,律齐,可闻及Ⅱ/Ⅵ级心脏杂音。腹膨隆,腹壁肿胀,无腹壁静脉曲张,肝右肋下2cm,脾脏肋下未触及,无移动性浊音,双下肢肿胀压之凹陷,会阴部肿胀。实验室检查:当地医院实验室检查:(1)尿蛋白3+,潜血2+ ;(2)血生化:ALB 17.5g,肾功无明显异常。(3)腹部彩超:肝胆胰脾双肾及腹腔未见明显异常。(4)胸片:右肺渗出性病变。入院后实验室检查:(1)尿液:黄色清亮,潜血1+,蛋白质3+,微量白蛋白定性(+)。(2)入院时查:总蛋白27g/L,白蛋白13.1g/L,血Ca2+1.62mmol/L 。胆固醇7.79mmol/L,甘油三酯4.05mmol/L。出院前查:总蛋白26.6g/L,白蛋白9.3g/L。(3)肌酐3.8μmol/L,尿微量蛋白浓度2.126g/L。(4)免疫Ⅰ号:免疫球蛋白G 0.884g/L,补体C3 0.48g/L,补体C4 0.11g/L。(5)HIV抗体、梅毒抗体、梅毒快速血浆反应素、结核抗体:阴性。(6)腹部彩超:①双肾形态结构及血供未见明显异常;②双侧输尿管未见明显扩张。(7)胸片:肺炎。(8)因患儿系新生儿,未行肾脏穿刺活检。
1.3 治疗及转归 患儿有大量蛋白尿,有严重低蛋白血症,最低时白蛋白<10g/L,水肿,高胆固醇血症。住院后反复给予人血白蛋白输注,并给予补钙、人免疫球蛋白等,经治疗患儿白蛋白未提升,还有下降趋势。入院治疗17d后,家长放弃治疗出院。
1.4 随访 出院时患儿仍存在水肿,出院前最近一次查白蛋白为9.3g/L,患儿于生后4个月左右死亡。
2 基因分析
患儿家系调查未发现家族中有类似疾病的成员。在知情同意情况下,采患儿、父、母的血完善先天性肾病相关基因检测:发现患儿存在NPHS1p.R1109X(c.3325C>T),NPHS1p.D310N(c.928G>A)(剪切区)复合杂合突变。如图1所示。
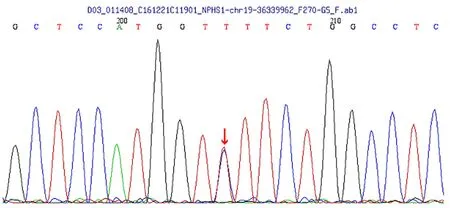
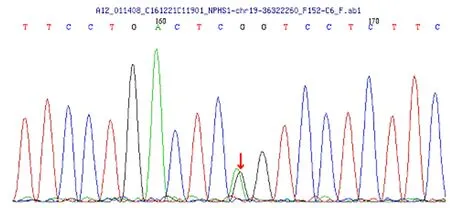
图1 患儿基因检测
发现患儿父亲NPHS1基因存在杂合突变,c.3325C>T,(编码区第3 325号核苷酸由胞嘧啶变异为胸腺嘧啶),导致氨基酸改变p.R1109X(无义突变),患儿母亲无该位点变异。该变异不属于多态性位点,在人群中发生频率极低。见图2。
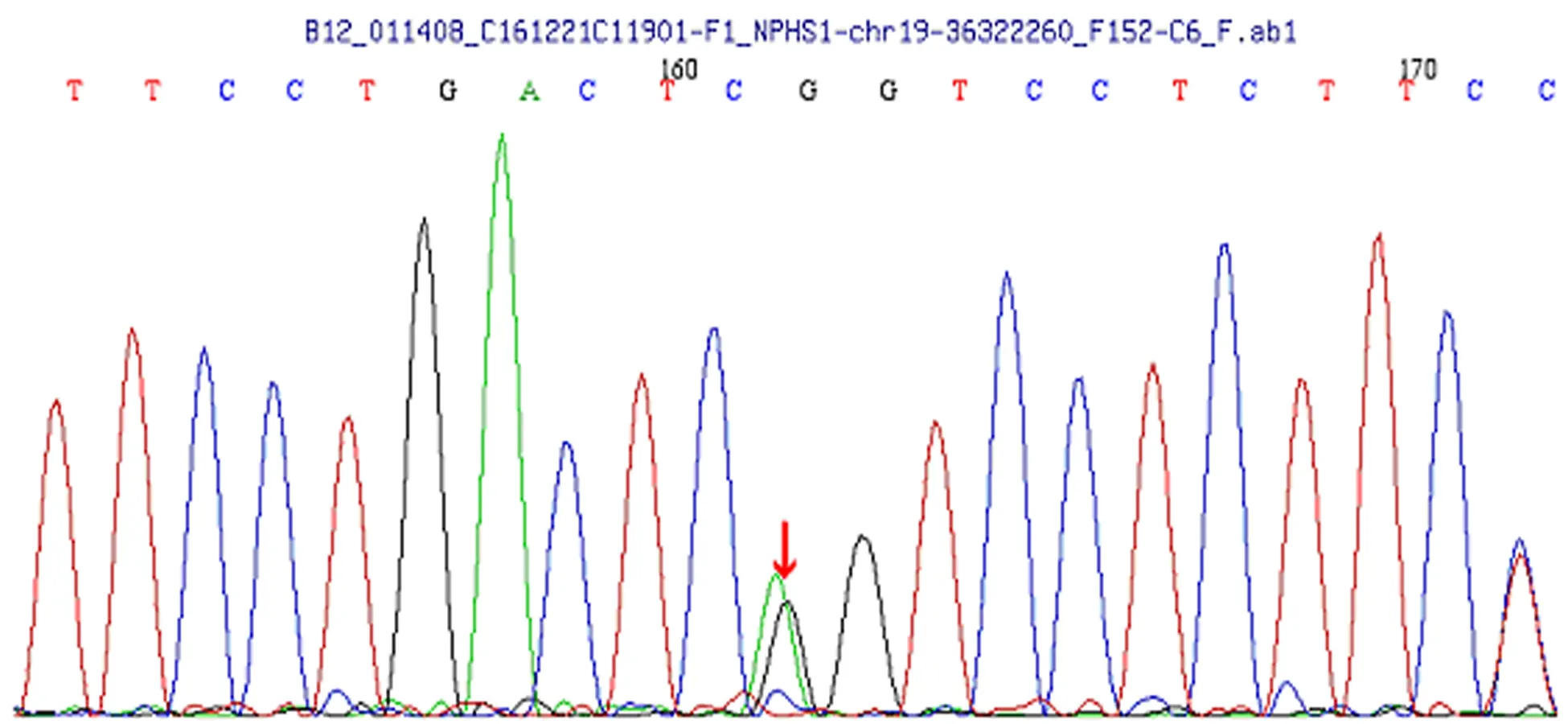
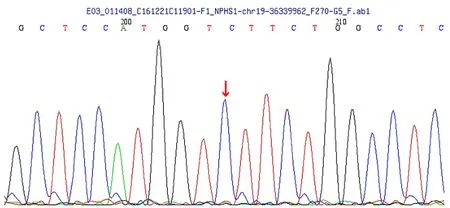
图2 患儿父亲基因检测
发现患儿母亲NPHS1基因存在错义突变,c.928G>A,(编码区第928号核苷酸由鸟嘌呤变异为腺嘌呤),导致氨基酸改变p.D310N(第310号氨基酸由天冬氨酸变异为天冬酰胺)。患儿父亲无该位点变异。该变异不属于多态性位点,在人群中发生频率极低。如图3所示。

图3 患儿母亲基因检测
3 讨论
3.1 CNS的分型和基因突变 CNS根据病因可分为原发性(遗传性)和继发性(非遗传性),继发性由多种病原体宫内感染或母亲疾病等导致,如梅毒、弓形体、巨细胞病毒、风疹病毒、肝炎病毒、人类免疫缺陷病毒以及疟疾等,除感染外,母亲SLE也可合并CNS[1]。原发性CNS由多个基因突变所致,目前已明确可以导致CNS的基因有NPHS1、NPHS2、WT1、PLCE1、LAMB2等[2],其中NPHS1、NPHS2编码蛋白多为肾小球裂孔膈膜蛋白分子,WT1、LMX1B编码蛋白为正常足细胞功能和发育所必需的转录因子或酶,LAMB2编码蛋白为肾小球基底膜结构分子[3]。本例患儿基因检测发现存在NPHS1基因杂合突变。本病发病机理为肾小球裂孔隔膜蛋白的编码基因NPHS1突变导致,导致足细胞足突间的裂孔隔膜受损,从而出现大量蛋白尿。
依据有无其他系统受累,又可分为非综合征型(non-syndromic)或单发型(isolated)和综合征型(syndromic),前者包括:Nephrin基因突变(NPHS1、芬兰型CNS)、Podocin基因突变(NPHS2)、WT1基因突变(单发型CNS)、PLCε1基因突变和LAMB2基因突变(单发型CNS);后者包括:WT1基因突变(Denys-Drash和Frasier综合征)、LAMB2基因突变(Pierson综合征)、LAMB3基因突变(赫茨交界性大疱性表皮松解症)、LMX1B基因突变(甲—髌综合征)、线粒体疾病(COQ2、COQ6)、CNS伴脑部畸形、Galloway Mowat综合征(基因不明)、CNS伴其他畸形(没有脑部畸形)和CNS伴心室扩大(基因不明)[4]。WT1基因突变为常染色体显性遗传,NPHS1、NPHS2、LAMB2和PLCε1等基因突变均为常染色体隐性遗传[5]。
国外、国内报道CNS较常见的基因突变为编码nephrin蛋白的NPHS1基因的突变,NPHS1基因突变导致的CNS综合征,临床上多指芬兰型CNS综合征,目前发现的突变位点数以百计,其中有2个典型突变:Fin-major(p.IJ41fsx91)和Finminor(p.Rll09x),94%的CNS病例有此两型突变,前者占78%,后者为16%[6]。
3.2 CNS的治疗 对于继发性CNS,主要针对其原发病治疗,如感染所致者,采用强有力的抗感染治疗,病情常明显好转,不出现不可逆性肾脏损害[7]。对原发性CNS,无特殊有效的治疗,类固醇激素或免疫抑制剂治疗无效。主要采取对症治疗及营养支持治疗。对于大多数原发性CNS患儿,肾移植是唯一有效的治疗方式。预后差,病情进行性发展,多死于感染、肾衰竭、出血等。
