HPLC测定盐酸苯达莫司汀有关物质与含量
2021-03-24孙朋杰卓秋琪
杜 超,孙朋杰,卓秋琪,唐 云,李 伟*
1.宜春学院(江西 宜春 336000) 2.深圳万乐药业有限公司(广东 深圳 518029)
盐酸苯达莫司汀是一种氮芥类抗肿瘤药物,具有烷基化和抗代谢活性双重作用。由于其安全性、耐受性和有效性,常被用于滤泡性淋巴瘤和其他低级别淋巴瘤的一线治疗。该药与其他烷基化剂相比,更易使DNA键断裂,在体内对抗癌细胞的时间更加持久;由于盐酸苯达莫司汀的结构中有3个活性基团,分别是烷化基团、丁酸侧链和苯并咪唑环。结构中丁酸侧链的引入,使苯达莫司汀具有酸碱两性的特质;烷基化基团使其具备氮芥家族的优势特点;苯并咪唑环使其具有抗代谢作用。其独特的结构使其具有烷基化和抗代谢双重治疗作用[1-2],盐酸苯达莫司汀结构式如图1。
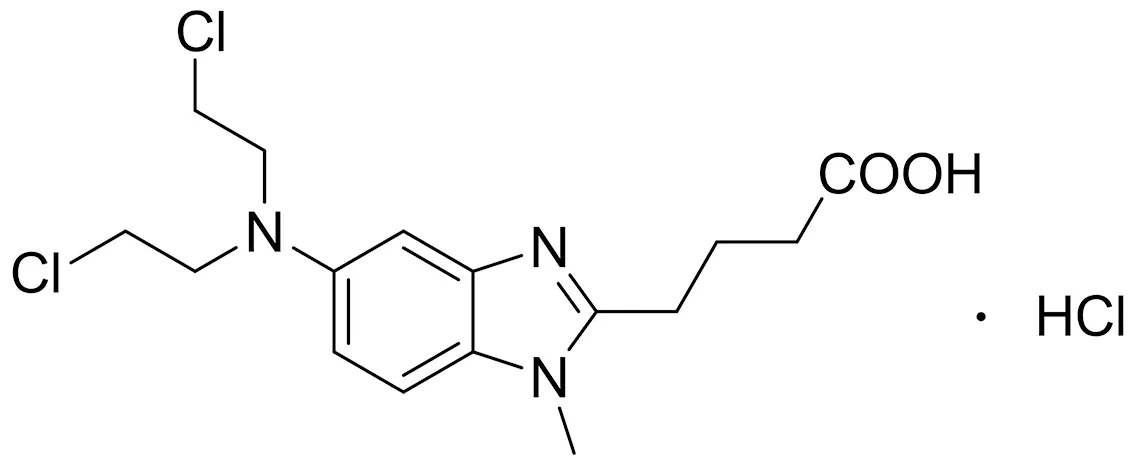
图1 盐酸苯达莫司汀结构式
目前,盐酸苯达莫司汀在国外已得到广泛的应用,盐酸苯达莫司汀在美国2008年被准予作为白血病的一线医治药物,国内多家医药公司对盐酸苯达莫司汀进行仿制与研究,但其使用与推广的程度却远远不够。同时对于其有关物质的控制与研究,也未有统一标准[3-9]。通过建立高效液相色谱法(HPLC)测定盐酸苯达莫司汀有关物质与含量,在后续的盐酸苯达莫司汀的研究工作中,将专利中[10-15]的梯度洗脱改为等度洗脱,在该条件下能够将盐酸苯达莫司汀生产和贮存中已知杂质,单烷基单羟基乙酯(杂质A),单羟基苯达莫司汀(杂质B),苯达莫司汀乙酯(杂质C)等进行有效分离。与专利方法比较,此研究方法能更好地解决各个杂质的归属问题,比以往的盐酸苯达莫司汀有关物质测定方法更具专属性,使有关物质与含量测定结果更加准确,能够更有效地控制药品的质量。
1 仪器与试药
1.1仪器高效液相色谱仪(Shimadzu Corporation,型号:LC-20AD(SPD-20A) ;电子天平(梅特勒-托利多国际有限公司,型号:XP205)。
1.2试剂与试药色谱级乙腈(Merck公司),色谱级甲醇(Merck公司),三氟醋酸(上海晶纯试剂有限公司),色谱三乙胺(TEDIA COMPANY.INC),磷酸(广州溢联化工有限公司),盐酸苯达莫司汀(深圳万乐药业有限公司,批号B19001、B19002、B19003),中间体BD-1(深圳万乐药业有限公司,190401),起始原料B0(深圳万乐药业有限公司批号:190313-1),苯达莫司汀对照品(F070G0,纯度99.6%,USP),中间体BD-1对照品(深圳万乐药业有限公司190401-2)。
2 方法与结果
2.1系统适应性与色谱条件色谱柱:Extend-C18,5 μm,4.6 mm×250 mm;流动相∶水-甲醇(36∶64)并含0.1% SDS、0.1%的三乙胺和0.1% H2PO4;等度洗脱;检测波长:233 nm;流速:1.2 mL/min;柱温:40 ℃。
从上述的系统适应图谱中可以看出,图2中主峰1与各有关物质以及主峰相邻峰都能够实现有效分离。主峰的保留时间为17.7 min,各个色谱峰之间的分离度为14.2、2.6、2.9、10.7、5.5、25.0、11.0、7.5。此条件下各色谱参数较为理想,因此初步将此液相条件确定为有关物质检查的液相条件。
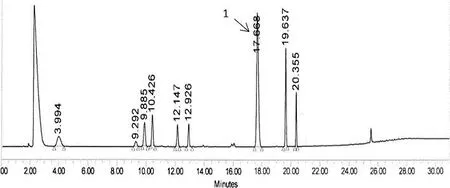
图2 盐酸苯达莫司汀系统适应性图
2.2溶液配制供试品溶液制备取盐酸苯达莫司汀供试品适量,精密称定,加甲醇溶解,定容并制备成1 mL中含0.5 mg盐酸苯达莫司汀的溶液。对照品溶液制备取盐酸苯达莫司汀对照品适量,精密称定,加甲醇溶解,定容并制备成1 mL中含0.5 mg盐酸苯达莫司汀的溶液。
2.3新建等度方法与原方法比较通过文献调研以及实验数据整理,将两种方法通过表格的形式进行比较,见表1。
2.4有关物质检测方法
2.4.1 方法专属性试验
2.4.1.1 破坏试验前样品溶液的测定 取盐酸苯达莫司汀(B19001)适量,按2.2项下溶液配制方法制备。
2.4.1.2 酸破坏 室温条件下,精密称取,盐酸苯达莫司汀样品(B19001)约12.5 mg,置25 mL量瓶。加盐酸溶液(1 mol/L)1 mL。将调配好的样品与空白溶液于60 ℃水浴放置1 h。水浴过后,取出样品与空白溶液并冷却至室温。之后,调节pH至中性,加溶剂定容至刻度,同法调配空白溶液pH至中性(防止剧烈条件对仪器损坏)。
2.4.1.3 碱破坏 室温条件下,精密称取,盐酸苯达莫司汀(B19001)约12.5 mg,置25 mL量瓶中。加氢氧化钠溶液(0.1 mol/L)2 mL,并按照同样的方法调配空白溶液。放置30 min,调节pH至中性,加溶剂定容至刻度,同法调配空白溶液pH至中性(防止剧烈条件对仪器损坏)。
2.4.1.4 光照破坏 室温条件下,取盐酸苯达莫司汀(B19001)适量,在日光灯10 000 lux下放置5 d,90 uW/cm2,1 d。2个循环以后,按2.2项下溶液配制方法制备。
2.4.1.5 高温破坏 室温条件下,取盐酸苯达莫司汀(B19001)适量,在130 ℃烘箱放置8 h后。冷却至室温,按2.2项下溶液配制方法制备。
2.4.1.6 氧化破坏 室温条件下,精密称取,盐酸苯达莫司汀样品(B19001)约12.5 mg,置25 mL容量瓶中。然后,加入30%过氧化氢浓溶液1 mL,浸润润湿后,并按照同样的方法调配空白溶液。之后,将调配好的样品与空白溶液,2.5 h后,加溶剂稀释至刻度。
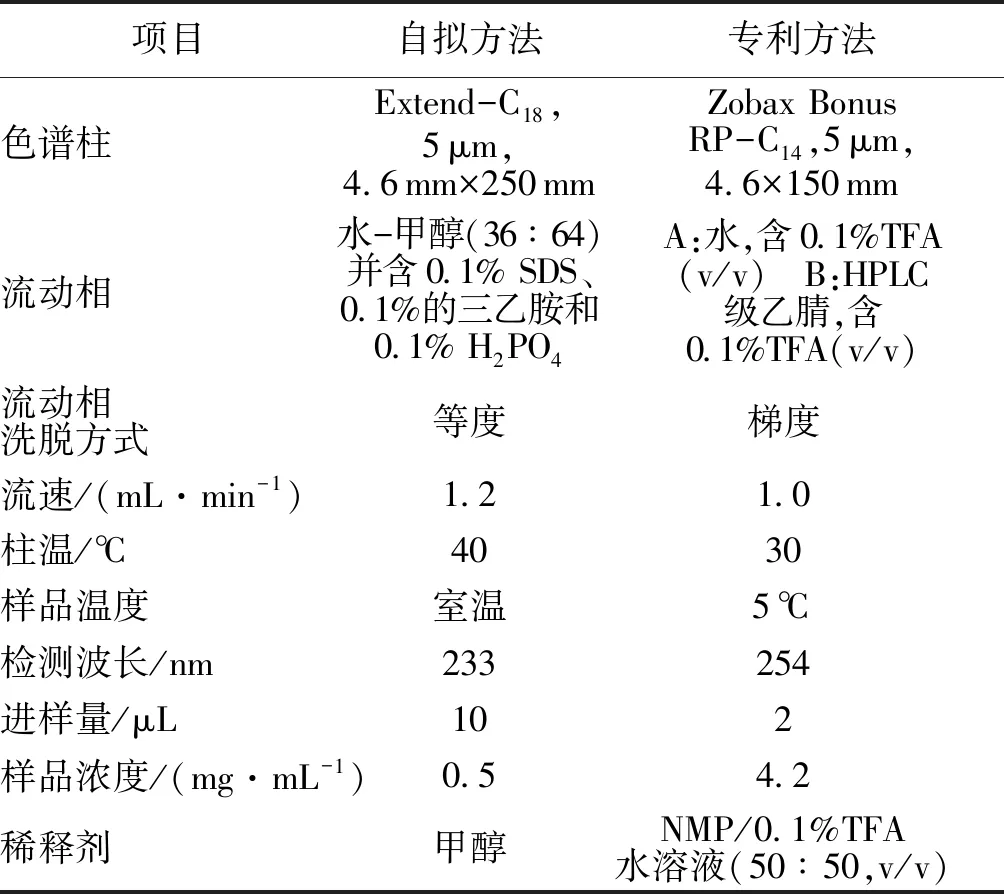
表1 新建等度方法与原方法比较
将上述样品、空白、破坏溶液,精密取10 μL,注入色谱仪。同时,考察了盐酸苯达莫司汀在不同的破坏条件下,主峰的成分与降解过程中杂质的相互分离作用情况。从实验数据可以看出,峰纯度较好,且破坏试验的质量平衡均在95%~105%范围内,质量平衡情况良好。通过高效液相色谱仪中的SPD-M20A检测器,对上述溶液的主成分峰,进行纯度检测,并结合各图谱,利用峰面积归一化法,得出各主峰的主成分均大于90%。综合上述破坏试验数据,可知该方法的专属性良好。破坏试验结果见图3、表2。
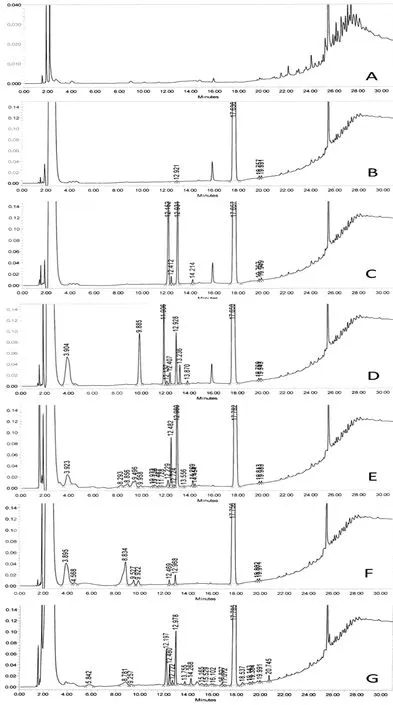
A:空白;B:样品;C:酸实验;D:碱实验;E:氧化实验;F:强光实验;G:高温实验图3 专属性试验色谱图
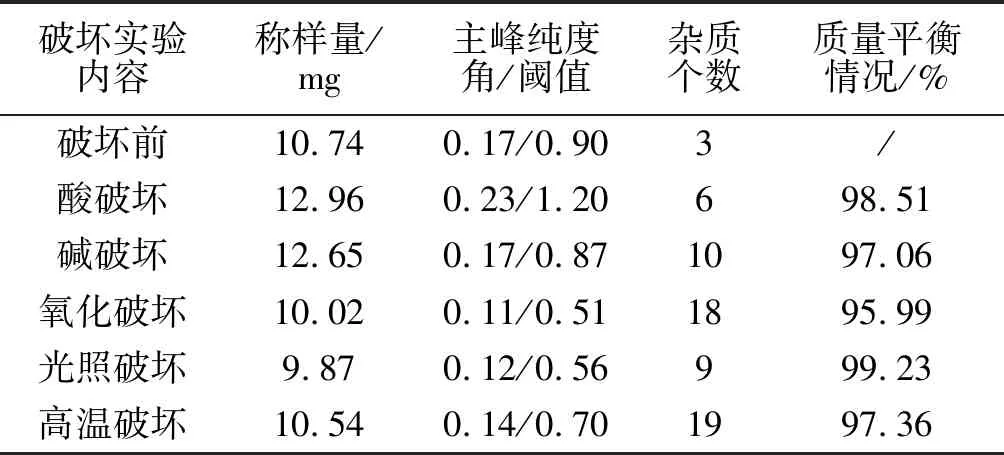
表2 破坏实验结
2.4.2 定量限与检测限 精密称取对照品适量,用甲醇溶解并稀释制成含盐酸苯达莫司汀0.82 mg/mL用稀释剂进行逐步稀释,当样品溶液的浓度为82 ng/mL时,进样10 μL,信噪比为10,故定量限为0.82 ng(n=6,RSD=1.6%);当样品溶液的浓度为24.6 ng/mL时,信噪比为3,故检测限为0.246 ng。
2.4.3 稳定性试验 取上述未破坏样品溶液,在0,2,4,6,8 h后,进样10 μL,记录色谱图。结果表明,盐酸苯达莫司汀的甲醇溶液在常温放置8 h,主峰峰面积RSD值为0.32%,说明主峰峰面积无明显变化,溶液稳定性较好。
2.4.4 样品有关物质的重复性 取盐酸苯达莫司汀(B19001)适量,精密称定,称重分别为10.0、10.2、11.3、11.1、10.4、10.1 mg。之后,按“2.2”项下溶液配制方法进行制备。进样10 μL,记录数据,结果表明,此法的重复性良好,见表3。
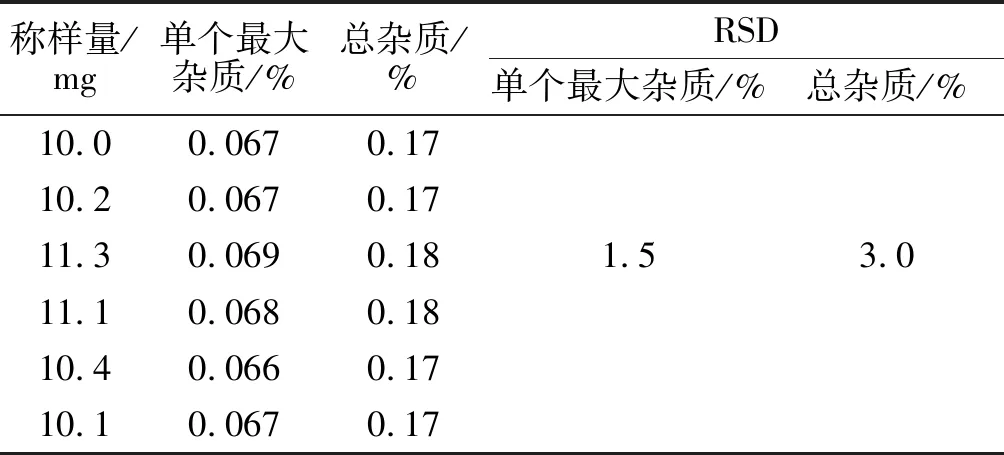
表3 重复性实验结果
2.4.5 色谱系统的耐用性
2.4.5.1 柱温的变动 将原始色谱条件中的柱温设定为(40±2)℃,对样品进行测定,记录数据,见表4。
2.4.5.2 流速的变动 将原始色谱条件中的流速调为(1.2±0.1)mL/min,对样品进行测定,记录数据,见表4。
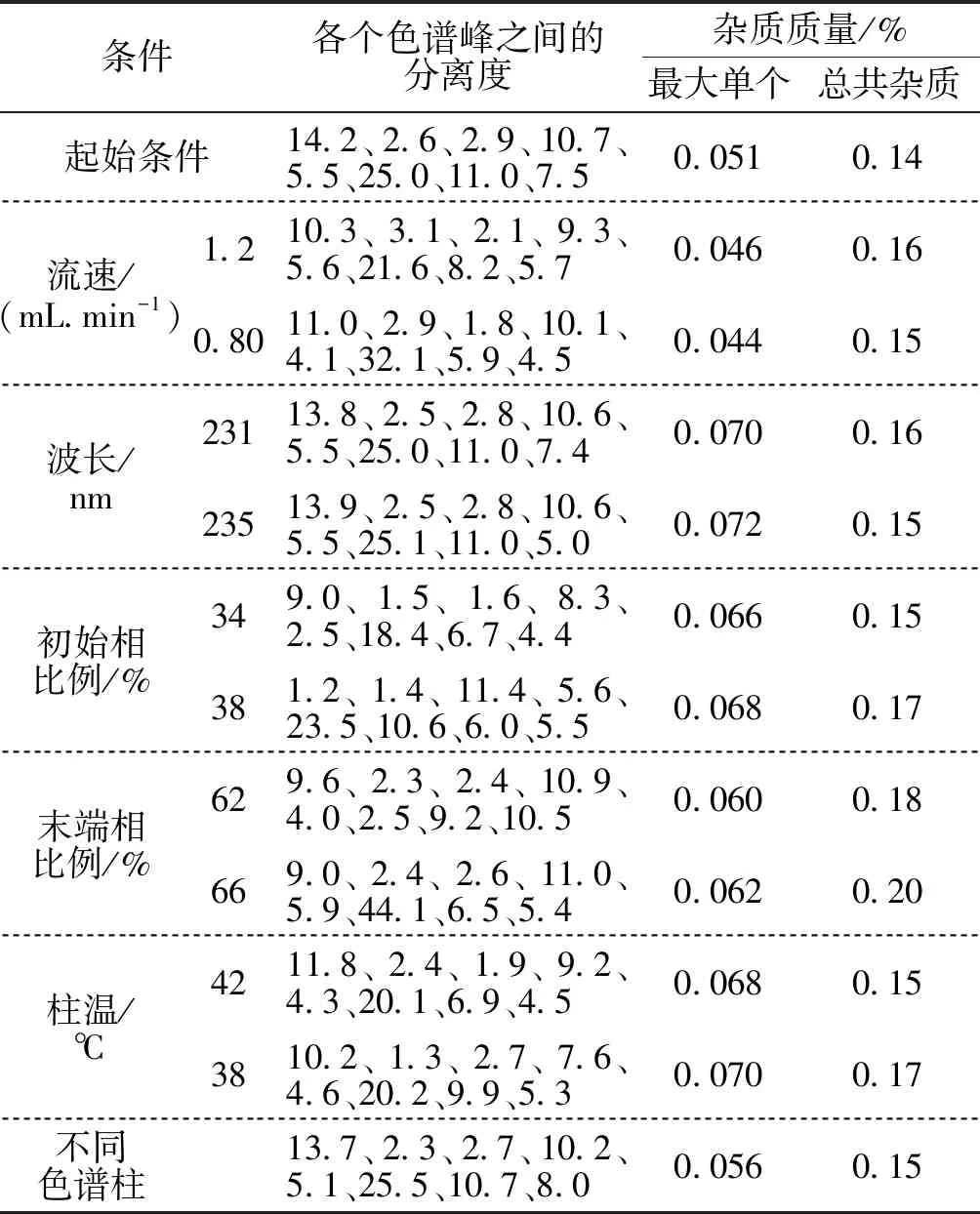
表4 耐用性数据总结
2.4.5.3 波长的变动 将原始色谱条件中的紫外检测波长调至(233±3)nm,对样品进行测定,记录数据,见表4。
2.4.5.4 选择不同品牌的C18色谱柱 将原始色谱条件中的色谱柱更换为DiamonsilTM(钻石)C18,5 μm,4.6 mm×250 mm,对样品进行测定,记录数据,见表4。
2.4.5.5 流动相成分的改变 将原始色谱条件中的初始水相比例调整为36%±2%,对样品进行测定,记录数据;将原始色谱条件中的梯度末端水相最高比例调整为64%±2%,对样品进行测定,记录数据。当设定的色谱条件发生细微改动时,各杂质相对保留时间基本稳定,各杂质的检出量未发生明显变化,且各杂质分离良好证明该法耐用性良好,见表4。
2.5含量测定方法
2.5.1 标准曲线的绘制 取对照品适量,按“2.2”项下方法制备成1 mL 中含盐酸苯达莫司汀0.04、0.06、0.08、0.10、0.12、0.14 mg的溶液,依法测定。
盐酸苯达莫司汀浓度与峰面积的线性方程为:y=3.916E+07x+9 066.2 (n=6),r=0.999 7。结果表明:在0.036 5~0.146 1 mg/mL浓度范围内,线性关系良好。
2.5.2 仪器精密度试验 取对照品适量,精密称定,按“2.2”项下方法制备。重复6次进样,得到RSD值为0.49%,证明该方法进样精密度良好。
2.5.3 重现性考察 取样品6份,按“2.2”项下方法制备,依法测定。6次进样,得到RSD=0.3%,平均含量为100.46%,证明该法重现性良好。
2.5.4 回收率试验 取样品6份,按“2.2”项下方法分别配制浓度为80%、100%、120%的低、中、高溶液3份,依法测定,平均回收率100.45%,RSD=0.4%,证明本法准确度较好,故此法适合本品的含量测定,见表5。
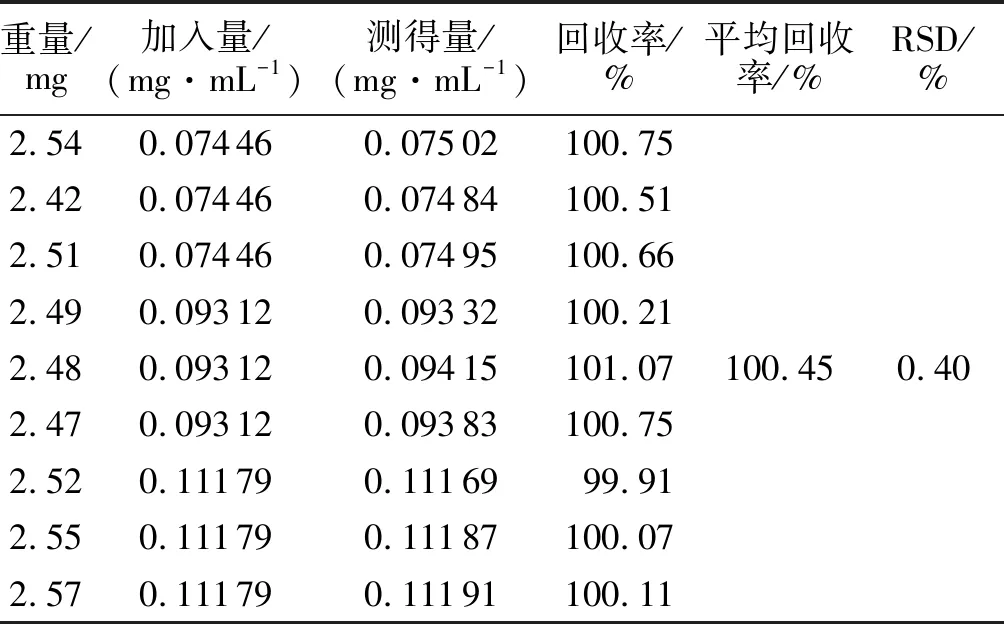
表5 盐酸苯达莫司汀回收率试验结果
2.6样品的有关物质及含量测定按照“2.1”项的色谱条件进行检测,结果见表6。该方法检测出的样品单个最大杂质,总杂质以及样品含量都符合规定,故该法适用于盐酸苯达莫司汀的有关物质以及含量测定。
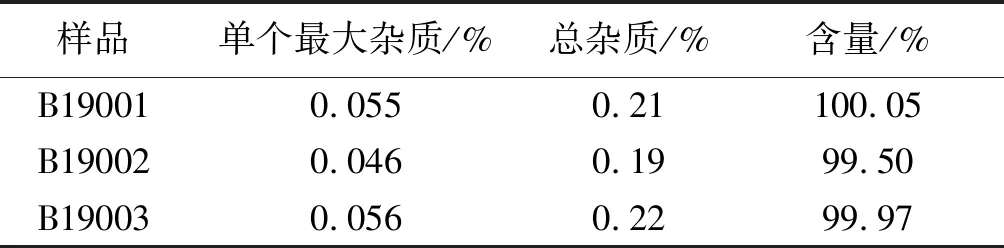
表6 盐酸苯达莫司汀有关物质及含量测定结果
3 讨论
3.1稀释剂的选择根据相关专利[10-15]报道,盐酸苯达莫司汀在水中迅速降解,随着醇浓度的增加,盐酸苯达莫司汀的稳定性也随之提高。根据上述情况,考察了本品在不同溶剂水、流动相和甲醇中的稳定性。
结果得知,本品在上述3种不同溶液中均无杂质C的产生。在水中因水的水解作用迅速降解,产生杂质A、B。本品在流动相比在水中稳定,降解杂质B只有少量的增加。本品在甲醇溶液中8 h内,杂质B的含量无明显变化,单个最大杂质和总杂质稍有增加,相对水和流动相,本品的甲醇溶液较稳定,这一数据和专利中溶液稳定性结果一致。同时,专利中的稀释剂NMP/0.1%TFA水溶液(50∶50,v/v),会导致溶液在放置过程中出现不稳定的现象,需临用新制,所以本研究选择甲醇做溶剂。
3.2检测波长的确定分别取起始原料、中间体、原料三批适量,按溶液配制法制备,进行紫外色谱扫描。鉴于三批样品、中间体及其各杂质在233 nm均有吸收,且检测基线噪音较小,从而表明在该波长检测,可得到良好的分离效果,故选择233 nm作为检测波长。
3.3色谱柱的选择本实验采用DiamonsilTM(钻石)C18,Extent C18等型号的色谱柱,在对盐酸苯达莫司汀进行测定时,从色谱图中可以看出,各峰的分离度、相对保留的时间、理论层数均可以保证实验结果符合要求,从而充分表明该实验方法的理论重复性良好。而且,使用Extent C18柱时,各峰理论塔板数相对较高,对称性较好,故采用Extent C18柱。
3.4进样量与样品浓度的选择因为专利文献方法中样品的浓度较大,如果在配制过程中,质量稍有增加,则色谱柱就很容易超载,出现平头峰的情况。所以,为了给后期的质量分析提供可靠的参考。将样品的浓度调整为0.5 mg/mL,进样量调整为10 μL。
3.5流动相的选择起初,在盐酸苯达莫司汀预实验处理时,流动相是水∶甲醇(36∶64)。但是,所得出的图谱出现主峰拖尾、峰形差,以及主峰保留时间位置过于靠前的问题。所以,在水:甲醇(36∶64)的基础上加入适量的三乙胺和磷酸以改善主峰拖尾的现象。同时,加入适量的SDS,得到合适的主峰保留时间。
3.6测定方法评价①按拟定的色谱条件连续进样6次,测得主成分结果RSD=0.49%,精密度良好。②8 h内主成分峰面积的RSD=0.32%,则证明在8 h内该系统适用性溶液较稳定。③经过线性回归,浓度约在0.036 5~0.146 1 mg/mL时,r=0.999 7,呈现良好的线性关系。④各批样品主成分回收率均处于98%~102%的范围内,回收率RSD=0.4%,回收率结果良好。⑤当设定的色谱条件发生细微改动时,各杂质相对保留时间基本稳定,各杂质的检出量未发生明显变化,且各杂质分离良好证明该法耐用性良好。⑥盐酸苯达莫司汀主峰可与各有关物质和降解杂质实现有效分离,且各主峰的主成分均大于90%。结果证明,该方法具有很好的专属性。⑦通过对同一份样品的多次测定,得到样品的单个最大杂质和总杂质数量都维持一定水平,则说明该方法重复性良好。⑧通过样品测定,表明各批样品有关物质和含量检测合格,符合要求。
综上所述,证明本研究拟定的色谱条件,简单,耐用,可靠。
