由双咪唑基配体和5-硝基间苯二甲酸构筑的二维镉(II)配位聚合物的合成和表征研究
2021-03-19朱倩倩段靖瑜董桂英
朱倩倩,李 薇,段靖瑜, 董桂英
(华北理工大学,河北 唐山 063210)
近年来,利用过渡金属和有机多齿配体构建新型金属-有机配位聚合物(MOCPs)引起了人们的广泛关注。这主要是由于它们有趣的构造、拓扑结构及其在导电、催化、吸附、磁性、荧光、多孔材料等领域的应用前景广阔[1-3]。然而合理地制备和预测特定配合物产品的确切结构和组成仍然是一个巨大的挑战。选择具有突出协作能力,功能和灵活性的金属中心和合适的桥接配体被认为是合理设计和合成MOCPs的关键因素[4-6]。众多学者致力于采用O-和N-供体有机配体,通过自组装桥接金属离子,形成具有无限网络结构[7]的MOCP。与此同时,混合配体自组装策略已逐渐被证实是有效的制备MOCPs的方法[8-9]。
1,3-双(咪唑基-1-甲基)苯(bix)是一种典型的柔性苯并咪唑衍生物,它能够满足金属中心的配位需求,从而产生更加强大和复杂的网络[10-12]。然而关于5-硝基间苯二甲酸构筑的MOCPs仍少见报道。因此,本文采用1,3-双(咪唑-1-甲基)苯(bix),5-硝基间苯二甲酸(H2nip)以及醋酸镉水热合成了一种新的二维网状配合物,{[Cd(bix)(nip)·H2O]·0.25H2O}n,并通过X-射线单晶衍射、热重(TGA)、红外光谱(FT-IR)对其结构及性能进行了表征分析。
1 实验部分
1.1 试剂和仪器
所有通过商业途径购买的化学药品直接使用,没有进一步纯化处理。使用Perkin-Elmer240C元素分析仪对C,H和N等元素进行分析;Nicolet FT-IR Avatar360分光光度计在4000-400cm-1范围扫描并收集了配合物的红外光谱;D/MAX 2500PC X射线衍射仪采用了Cu-Kα射线进行了X射线粉末衍射;用Hitachi F-7000荧光分光光度计在固态下于室温收集了相关数据;热重分析则是在Netzsch TG209热分析仪上于氮气气氛下,以10℃/min的升温速率从室温到800℃收集了相关数据。
1.2 {[Cd(bix)(nip)·H2O]·0.25H2O}n的合成
将Cadmium acetate dihydrate(0.1mmol,26.60mg),H2nip(0.1mmol,21.10mg)和含氮配体bix(0.1mmol,37.80mg)溶解于蒸馏水(10mL),并不断搅拌30min。然后将混合溶液密封在25 mL聚四氟乙烯内衬的不锈钢反应釜中,以5℃/h的升温速度在140℃下反应72h。得到无色块状透明晶体,以Cd为基础产率为53%。所得晶体的分子式为C22H19.50CdN5O7.25(分子量为582.33)。元素分析结果(括号内为理论值)分别如下:C,45.02%(45.38 %);H,3.08%(3.38%);N,12.28%(12.03%)。主要红外光谱数据(cm-1):3418(vs),3125(s),1616(vs),1562(s),1526(s),1451(m),1361(vs),1344(vs),1230(m),1109(m),1088(m),724(vs),654(m).
1.3 X-射线晶体衍射
配合物的晶体学数据是通过Bruker Smart 1000 CCD衍射仪以Mo-Kα辐射(λ= 0.71073Å)在293(2)K下使用ω-2θ扫描模式收集的,分别使用SADABS多扫描程序和SHELXS程序用直接法进行吸收校正与晶体结构解析,并使用SHELXTL程序在F2水平上通过全矩阵最小二乘法对非氢原子的坐标和各向异性的温度因子进行细化[13-14],水分子中的氢原子由差值傅里叶合成确定,其它有机物上的氢原子理论加氢确定。图形处理在Diamond软件中完成。表1提供了配合物的详细晶体学数据和结构优化参数。表2提供了配合物的选定键长和键角。

表1 配合物的晶体学参数
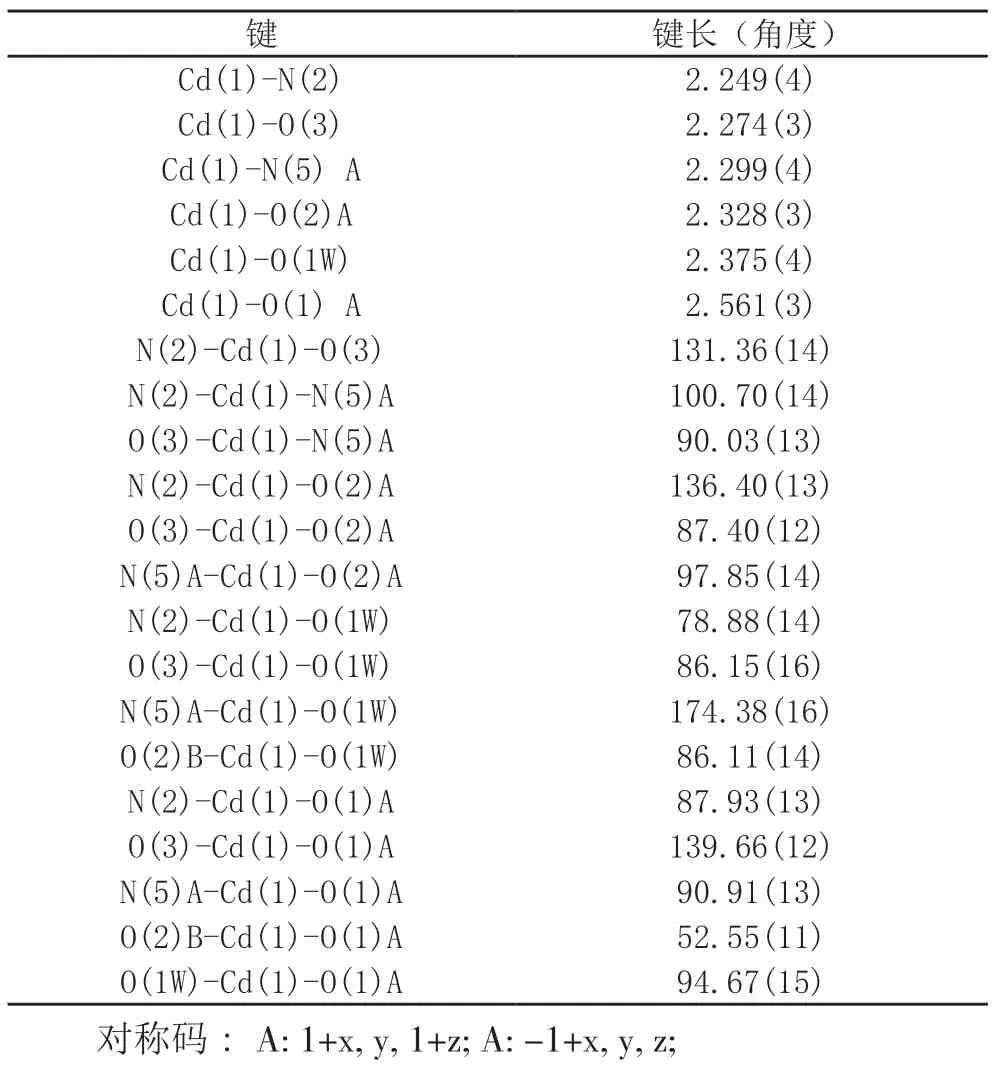
表2 配合物选定键长(Å)和角度(°)
2 实验结果与分析
2.1 晶体结构描述
X射线单晶衍射分析表明,配合物的最小不对称单元包括一个晶体学独立的Cd(II)中心,一个bix配体,一个nip2-配体,一个结合水分子和0.25个晶格水分子。如图1所示,Cd(II)采用六配位模式,与两个来自不同bix配体的N原子(N2,N5B)、结合水中的一个氧原子(O1W)和来自两个不同nip2-配体螯合羧酸中的两个氧原子、一个单齿羧酸中的氧原子(O1A, O2A,O3,对称代码:A:x+1,y,z+1; B:2=x-1, y,z)相连构成了一个稍微扭曲的八面体几何构型。O1A,O2A,O3和N2构成赤道面,轴向位置由O1W和N5B占据,O1W-Cd1-N5的角度为174.38(1)°,Cd-N键长分别为2.249(4)和2.299(4)Å,Cd-O键长为2.274(3)到2.561(3)Å不等,均在正常范围内[15-16]。

图1 配合物的配位环境图
去质子化的nip2-配体中两个羧酸基团采用μ2-η1, η1配位模式,nip2-配体连接两个Cd(II)形成一维线性[Cd(nip)]n链,Cd…Cd 距离为10.328(1)Å(图2)。值得注意的是,每个bix配体连接两个Cd(II)离子,每个Cd(II)离子连接两个bix配体,通过bix配体连接的Cd-Cd的距离为11.314(2)Å。通过bix配体可以将一维[Cd(nip)]n链在ac平面进一步拓展成为平行四边形网格状结构。每个网格状单元都由四个分散在顶角的Cd(II)原子通过连接两个bix和nip2-配体构成的四边组成(图3)。为进一步了解结构,对聚合物进行了拓扑分析,由TOPOS4.0程序计算,Cd(II)离子可视为4-连接节点,bix和nip2-配体视为连接体,因此配合物的结构可以被认为是一个单节点的4连接非互穿sql四方平面网,点符号为44.62的拓扑结构。此外,配位水分子O1W与游离水分子O2W与羧酸氧原子O3之间存在两个O-H…O氢键相互作用(O…O =2.701(5)~3.026(4)Å和∠O-H…O=161(7)°),进一步稳定了配合物的晶体堆积(表3)。

图2 配合物一维链状结构图
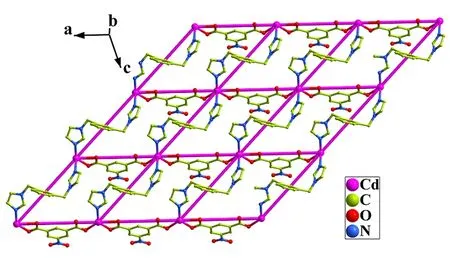
图3 配合物的二维网状结构图

表3 配合物的氢键几何形状(Å)和角度(°)
2.2 热重分析
为研究配合物的骨架稳定性,进行了热重(TG)分析,TG曲线如图4所示。分析TG曲线可知,在29~113℃范围内,由于配位水分子和晶格水分子的脱出配合物第一次失重4.53%(理论值:4.29%)。在113℃之后,配合物质量保持稳定,直到327~773℃有机配体bix和nip2-分解,配合物骨架坍塌,导致第二次失重74.59%(理论值:76.83%)。在820℃时获得煅烧残渣,推定为CdO(重量占比22.28%,理论值:22.05%)。
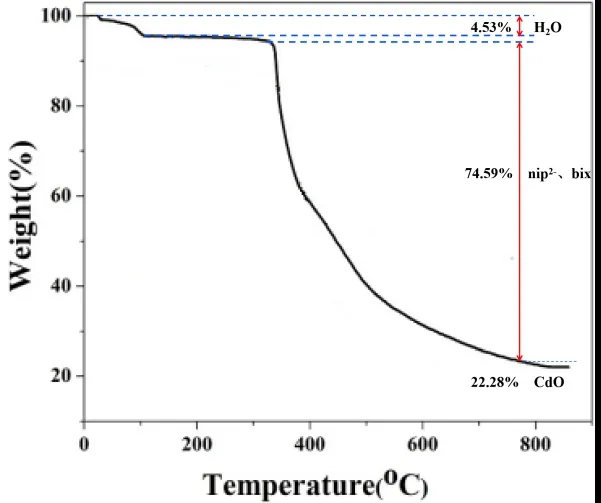
图4 配合物的热重曲线图
2.3 红外光谱及XRD图谱分析
配合物在3674~3184cm-1范围内发现一个属于配位水分子O-H键伸缩振动的强宽吸收带,这表明配合物中存在水分子。同时,bix配体咪唑环中的νC=N使得配合物在1526cm-1处出现吸收峰。反对称伸缩振动νas(COO-)在配合物中引起1616和1562cm-1的强峰,对称伸缩振动νs(COO-)在配合物中引起1451和1361cm-1的强峰。此外,配合物的Δν(Δν=νas(COO)-νs(COO)])值为165和201cm-1,说明nip2-配体中的羧基采用单齿和双齿鳌和两种不同的模式。
为了确定粉末样品的相纯度,对化合物进行了X射线粉末衍射(XRPD)实验(图5)。可以看出,除了强度的差异,单晶X射线衍射和实验模拟的峰在关键位置上很好地匹配,这也证实了每个化合物样品的相纯度。

图5 配合物的XRD图谱
3.总结
在水热条件下合成了一种含有柔性双咪唑和5-硝基间苯二甲酸配体的镉(II)配合物。配合物所属晶系为单斜晶系,空间群为P 21/c,拓扑为二二维(4,4)网络,通过对其进行的热分析可知该配合物有较高的热稳定性。
