原发性开角型青光眼大家系遗传眼病多基因测序结果分析
2021-03-18叶龙玲
周 瑾,姚 磊,刘 君,叶龙玲,张 毅,陈 谨*
(1.安徽医学高等专科学校,合肥 230601;2.合肥名人眼科医院,合肥 230601;3.金域医学检验中心,合肥 230601)
原发性开角型青光眼(primary open-angle glaucoma,POAG)是一类遗传性致盲眼病,其病因和发病机制还不十分清楚,但其遗传性在发病中的作用已引起了广泛的重视。大量研究显示,青光眼是一种多基因遗传性疾病,已有的诊断和治疗方法存在着极大局限性。与青光眼有关的致病基因包括OPA1、MYOC、CYP1B1、OPTN等〔1-4〕,但这些已报道的基因暂不能完全导致所有青光眼的发病,肯定还存在其他与青光眼遗传性因素相关的基因位点或基因尚未被发现。因此,选取具有典型临床意义的青光眼患者及其家系,进行基因遗传学研究,具有十分重要的价值〔5-6〕。
1 标本收集
收集合肥名人眼科医院开角型青光眼患者及其家系成员(父亲、母亲、妻子、女儿、亲妹妹、堂兄弟、堂兄弟妻子及堂兄弟儿子)全血标本9 例,进行遗传眼病的基因测序分析。在经患者及家系其他成员的知情同意后,选择家系的1名POAG 患者和8名正常成员的外周血DNA,对遗传性眼病相关的404个基因进行测序,分析测序数据。
2 患者临床资料
患者,男,34岁,系“双眼抗青光眼术后10年,左眼胀视物不清2 月”入院,患者于10 年前曾在合肥名人眼科医院行双眼小梁切除加虹膜周切术,手术顺利,病情平稳出院,2 月前来院复查,无明显诱因下出现左眼胀伴视物不清,无恶心、呕吐等症状,现眼压控制不理想,为求诊治,门诊拟“左眼开角青光眼术后眼压失控,右眼开角型青光眼术后”收入院。既往史:2008 年曾在淮南某医院行双眼准分子激光手术,2009 年5 月20 日 及2009 年5 月28 日曾 在 合肥名人眼科医院分别行双眼抗青光眼手术,2015 年4 月曾行右小梁切除加虹膜周切术。否认肝炎、肺结核等传染性疾病史,否认输血史,否认药物及食物过敏史,预防接种史不详。
眼科检查见图1~2。裸眼视力:右眼(OD)0.5,左眼(OS)颞侧眼前指数(戴)。右眼睑肤色正常,无睑内外翻,无倒睫,未扪及肿块,启闭自如,球结膜充血(+),角膜通明,前房深浅正常,房闪(-),虹膜上方1 点及11 点方向可见两处根切口,瞳孔圆,直径约4 mm,对光反应迟钝,晶体混浊,玻璃体混浊,眼底视盘色苍白,杯盘比=0.90;左眼眼睑肤色正常,无睑内外翻,无倒睫,未扪及肿块,启闭自如,球结膜充血(++),上方滤过泡扁平隆起,切口对合可,缝线在位,角膜透明,前房浅,上方1 点方向可见引流钉植入,膜上方12 点方向可见根切口,瞳孔圆,直径约2 mm,呈药物性缩小,晶体混浊,玻璃体混浊,眼底视盘色苍白,杯盘比=0.95;眼压R∕L(右∕左)=21.2∕9.0 mmHg(1 mmHg=0.133 3 kPa)。
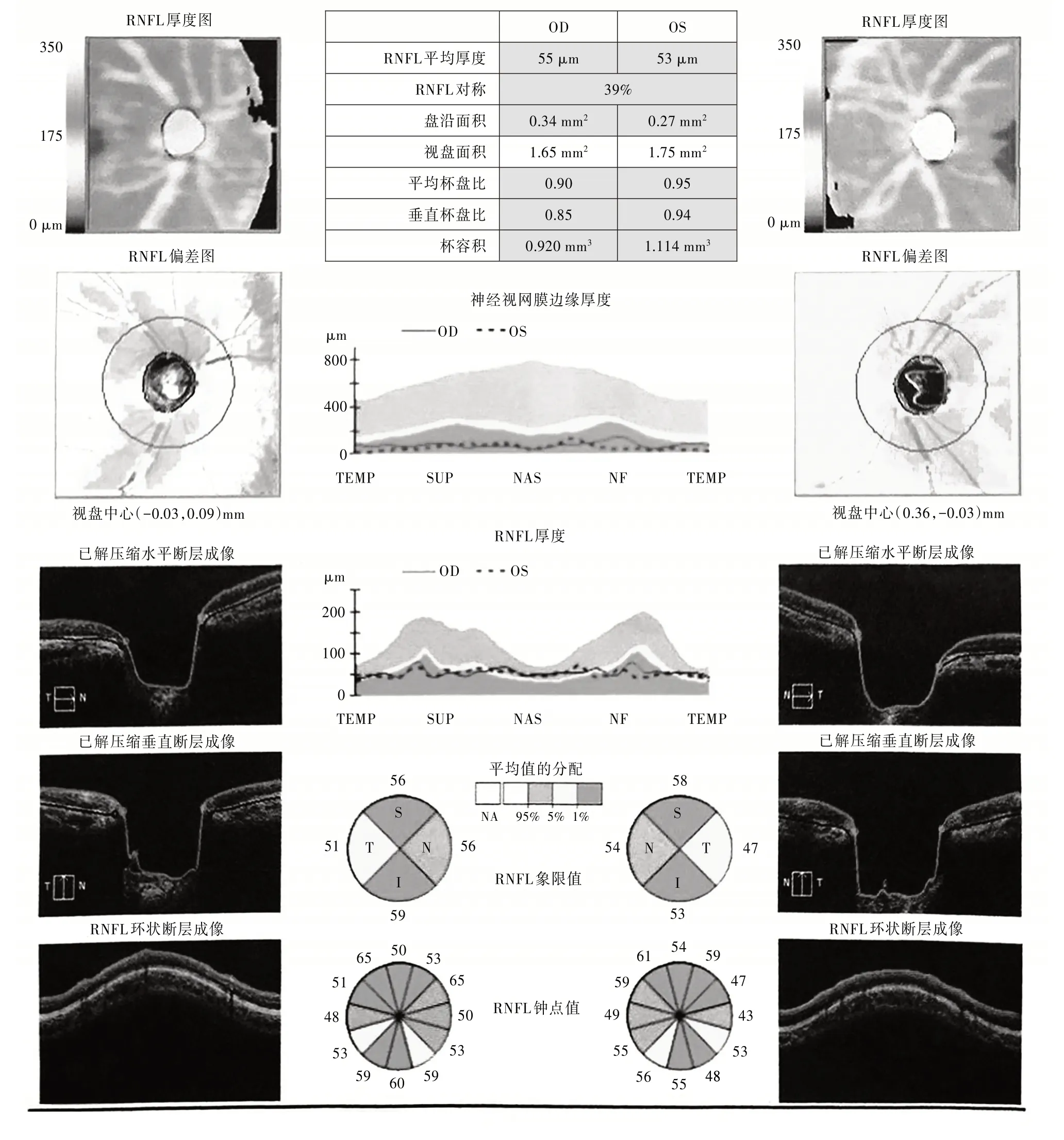
图1 双眼神经视网膜RNFL(视网膜神经纤维层)和ONH OU(视神经乳队双眼)分析
其他受检者未出现视力下降、眼胀痛等眼部不适的症状,同时行眼科检查,裂隙灯检查前房角正常,眼压正常,眼底无异常,视野检查均正常。排除POAG。
3 基因检测结果
通过与金域医学检验中心合作,检测涉及404个基因,其中患者及其女儿有两个相同的基因突变位点,分别是RPGRIP1基因,染色体位置14q11,参考序列NM_020366.3,第6 外显子,cDNA 水平的844 位碱基C 变成碱基T,使得蛋白水平的282 位氨基酸由亮氨酸(Leu)变成苯丙氨酸(Phe),发生Leu282Phe杂合突变;OPA1基因,染色体位置3q29,参考序列NM_015560.2,第2 外显子,cDNA 水平的320 位碱基C 变成碱基T,使得蛋白水平的107 位氨基酸丝氨酸(Ser)变成Leu,发生Ser107Leu 杂合突变。以上变异均为错义突变。见表1。
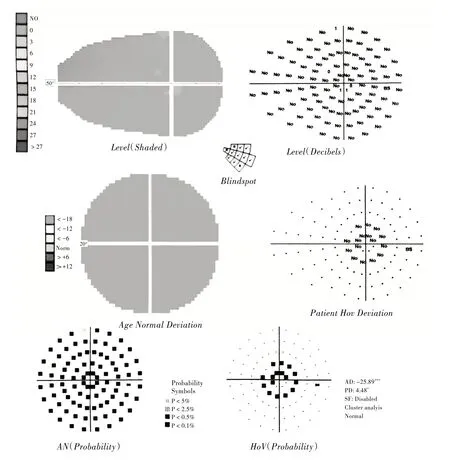
图2 患者右眼视野

表1 患者眼部突变基因检测结果
Leu282Phe 错义突变使所编码的蛋白质第282位氨基酸由Leu 变为Phe。见图3。HGMD 数据库未见文献报道,ESP6500siv2_ALL 和千人基因组(1000 g 2015 aug_ALL)数据库未见收录,dbSNP147数据库有收录。Ser107Leu错义突变使所编码的蛋白质第107位氨基酸由Ser变为Leu。见图4。HGMD 数据库未见文献报道,千人基因组(1000 g 2015 aug_ALL)数据库未见收录,ESP6500siv2_ALL 和dbSNP147 数据库有收录(rs781398129)。生物信息学软件预测上述两个突变有致病可能性。
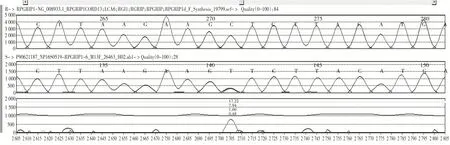
图3 RPGR1P1错义突变(Leu282Phe杂合突变)
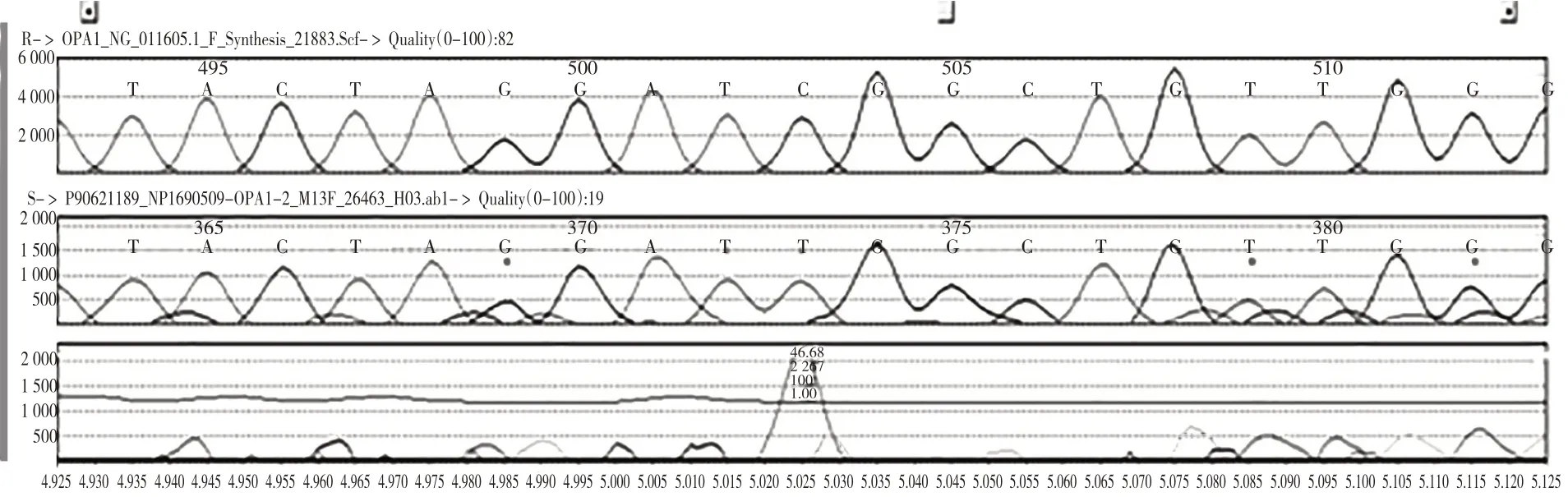
图4 OPA1错义突变(Ser107Leu杂合突变)
患者的女儿(9 岁)是两个错义突变的携带者,却没有明显的临床症状,属于正常眼,建议长期、定期监测双眼视力变化。
患者的其他7位家属均未检测到遗传性眼病相关基因外显子编码区的致病性变异。阴性结果降低了患者存在已知致病基因变异的可能性,但由于该疾病的临床表现及相关基因变异类型均存在高度多样性,通过现有的基因检测技术难以检测到所有类型的变异。从现有检测结果可知,本方法的可检测范围内没有任何的致病性突变,但还应注意的是,几乎所有基因检测方法都会存在某些局限性,尤其是当临床上十分疑似的病例却没有检测到致病性变异时,本研究中可能与患者女儿年纪较小,尚未发病有关。
4 结论
检测到POAG 患者RPGRIP1基因的Leu282Phe杂合突变和OPA1基因的Ser107Leu 杂合突变,考虑可能与POAG 发病有关,但其女儿(9 岁)同样携带RPGRIP1基因的Leu282Phe 杂合突变和OPA1基因的Ser107Leu 杂合突变,暂时未发病。推测可能存在目前尚不知道的POAG 致病基因,或已知眼病相关致病基因并不能覆盖所有POAG 患者;同时考虑患者24 岁青光眼发病,需要长期监测本青光眼家系的视力、眼压、眼底杯盘比、视野和视网膜神经纤维厚度等,以期为基因治疗青光眼的进一步研究提供参考。
POAG是青光眼最常见的类型之一〔7〕,约占青光眼发病的50%。已有的研究显示,发达国家约50%的POAG 患者未被早期发现〔8〕,我国亦有约80%的POAG 患者未被早期发现〔9〕。因POAG患者早期多无明显临床症状,当临床症状逐渐明显时,多已进展到中晚期,其视野明显变窄,视力明显降低,进而出现不可逆的视神经损伤。因此,POAG 患者的早期诊断显得尤为重要。
POAG 作为一类遗传性致盲眼病,相关致病基因的研究和确立将有助于临床确诊患者,早期发现,早期治疗。通过对404 个遗传眼病的基因进行测序,为临床寻找有效的POAG 诊断和治疗提供新的途径和手段,同时检测与POAG 相关的更多位点与基因,为实现POAG的基因筛查、基因诊断及基因治疗提供理论支持。而基因的改变是否会引起患者的临床疾病,尚有待科学的进步与发展、数据的积累及更深入的基因功能学的研究。针对此类基因改变,临床及实验室需要密切关注临床进展,在适当时候对结果进行补充说明,同时拓展检测的基因种类。
