1⁃羟基色胺衍生物的合成研究
2021-03-15刘志波
赵 鹏,刘志波,于 芳
(辽宁石油化工大学石油化工学院,辽宁抚顺113001)
亲核反应作为有机化学中常见的反应,在现代有机合成中一直被广泛使用。亲核反应分为亲核加成反应和亲核取代反应,两种反应中都需要化学性质优良的亲核试剂。合成一种化学性质良好的亲核试剂一直是科研人员的研究重点。
近年来,随着生物碱研究的兴起,各类不同生物碱的医学及药学价值不断被开发出来。现代化学家希望通过化学合成的手段高效地合成这些生物碱。生物碱的种类繁多,其中吲哚生物碱占比很大。本文希望合成一种能够在制备吲哚生物碱过程中起到至关重要作用的亲核试剂。
2007年,M.Somei等[1]进行了1⁃羟基化合物的相关研究,如图1所示。研究发现,化合物1具有明显的特点,在吲哚环的1⁃位上存在着一个羟基,导致该化合物有较好的反应活性。作者利用化合物1反应活性高的特点,以化合物1为中间体,完成了化合物2的合成。
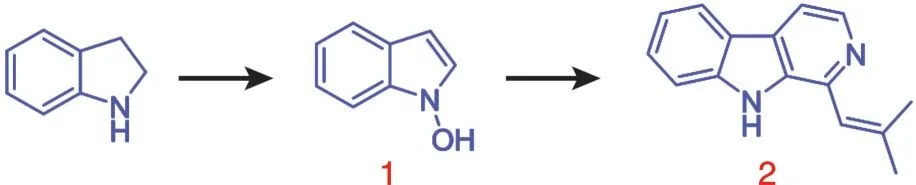
图1 化合物2的合成
2015年,M.Somei等[2]通过氧化化合物3形成1⁃羟基化合物,然后在重氮甲烷的作用下生成化合物4,如图2所示。M.Somei希望将化合物4经过一系列的转化,生成含有3⁃位偶联的两个6,5,5并环结构,但是,经过一系列的实验,并未得到化合物5。

图2 1⁃甲氧基色胺化合物的制备
2015年,Q.Wang课题组利用1⁃羟基化合物与苯炔反应得到了碳氮键偶联的产物[3],如图3所示。首先化合物6与苯炔反应得到中间体7,中间体7经过1,3⁃重排得到了碳氮键偶联的化合物8。

图3 1⁃羟基化合物与苯炔的反应
经过上述研究者对1⁃羟基化合物的制备及其在生物碱合成和偶联反应中的应用研究,相关科研人员意识到了1⁃羟基化合物的重要性。随着生物碱合成的兴起,将1⁃羟基色胺衍生物应用于吲哚生物碱的合成中具有重要意义。因此,本文进行了系统的探索及研究,高效地合成了一系列1⁃羟基色胺衍生物,希望这些衍生物能够在将来的吲哚生物碱合成中起到积极作用。
1 实验部分
1.1 实验药品及仪器
N,N⁃二甲基甲酰胺、三氯氧磷、硝基甲烷、氢化铝锂、氯甲酸甲酯、三乙基硅烷、二水合钨酸钠、吲哚、4⁃甲基吲哚、5⁃氯吲哚、6⁃甲基吲哚、7⁃溴吲哚、氢氧化钠、乙酰氯、二碳酸二叔丁酯、间氯过氧苯甲酸、二水合钨酸钠、过氧化氢、氰基硼氢化钠、三乙胺、甲醇、氯化钠、无水硫酸钠、碳酸氢钠、二氯甲烷、四氢呋喃、三氟乙酸、石油醚、乙酸乙酯,分析纯,萨恩化学技术(上海)有限公司。
Bruker Avance 500型超导核磁共振仪,瑞士Bruker公司;VARIAN 4000气相色谱/质谱联用仪,美国瓦里安技术中国有限公司;90⁃1磁力搅拌器,上海司乐仪器有限公司;HX⁃2015超级恒温循环泵,郑州长城仪器有限公司;X系列旋片式真空泵,淄博东圣真空设备有限公司;RE⁃2000B型旋转蒸发仪,巩义市予华仪器有限责任公司;DFY⁃5/40型低温恒温反应浴,郑州市亚荣仪器设备厂;SHZ⁃D(Ⅲ)循环水式真空泵,巩义市英峪予化仪器厂。
1.2 反应条件的优化
以取代的吲哚为起始底物,通过甲酰化反应、亨利反应和氢化铝锂还原,得到具有不同取代基的色胺类衍生物。取代的色胺与各种酰氯或酸酐反应生成侧链N取代的色胺衍生物,经过三乙基硅烷还原、过氧化氢氧化,以较高的收率得到1⁃羟基色胺类化合物[4],如图4所示。
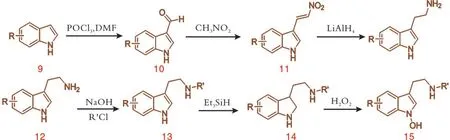
图4 化合物15的合成路线
化合物13经过还原生成化合物14。通过加入不同底物发现,现有条件不能满足所有化合物的合成。因为还原反应条件较为剧烈,如果侧链的氨基上有叔丁氧羰基保护,该化合物就会在三氟乙酸的条件下脱保护。因此,有叔丁氧羰基的底物应选择氰基硼氢化钠作为还原剂[5]。如图5所示,在乙酸做溶剂的条件下,含有叔丁氧羰基的化合物13h,经氰基硼氢化钠还原生成化合物14h。

图5 化合物14的合成路线
化合物14经双氧水(过氧化氢)氧化生成化合物15。在氧化反应的选取过程中,查阅文献[6]后,选择了两种方案进行氧化。
首先,在二氯甲烷作溶剂、间氯过氧苯甲酸做氧化剂的条件下,将化合物14氧化生成化合物15,如图6所示。反应12 h后,经过薄层色谱检测,发现少量原料剩余,生成的产物也较少。经过连续的薄层色谱检测和查阅文献[7],发现在反应过程中产物被氧化剂进一步氧化生成氮氧化物的副产物。经过大量实验发现,底物在该氧化条件下收率较低并且无法控制反应进程。

图6 间氯过氧苯甲酸的氧化
其次,在二水合钨酸钠作为催化剂、过氧化氢作为氧化剂的条件下,持续搅拌10 min,反应结束。在该反应条件下,0.1 mmol化合物14反应0.5 h,以80%的收率得到化合物15。但是,当化合物14的物质的量增加至1~10 mmol时,产物收率会明显下降(小于30%)。并且在反应物物质的量相同的条件下,将此反应重复进行,每次反应后得到的产物收率差距较大。因此,后续从反应温度、反应物浓度以及实验后处理等方面对反应进行研究。研究发现,体系内残余的过氧化氢在后处理的过程中将产物继续氧化,从而影响产物收率,因此对反应后处理操作进行优化。反应结束后,将反应液用水进行多次洗涤以除去残余的过氧化氢,并用淀粉碘化钾试纸检测,确保过氧化氢完全除去后,用饱和氯化钠水溶液、无水硫酸钠进行除水和干燥,经过柱层析分离,得到1⁃羟基色胺衍生物,收率为80%,并且该反应可以放大到克级进行制备。
1.3 操作步骤
化合物9a到化合物15a的合成路线见图7。

图7 化合物9a到化合物15a的合成路线合成路线
化合物9a经甲酰化反应生成化合物10a[8]。准备烘干的圆底烧瓶,将体系进行氩气保护。将N,N⁃二甲基甲酰胺加入到圆底烧瓶中(每1 mL三氯氧磷加5 mLN,N⁃二甲基甲酰胺),然后将体系置于0℃下缓慢地滴加2.5 mmol三氯氧磷。体系在0℃下搅拌5 min,然后将含有1.0 mmol化合物9a(每1 g的化合物9a加10 mL的N,N⁃二甲基甲酰胺)的N,N⁃二甲基甲酰胺溶液加入到反应体系中。将反应体系升至室温,剧烈搅拌3 h,以免因为体系黏稠而导致反应不均匀。搅拌完成后,向体系里加入2.6 mL 3.8 mol/L的氢氧化钾水溶液,搅拌均匀后加热回流过夜。反应完全后,加入饱和的碳酸氢钠水溶液和乙酸乙酯直至溶液变得澄清。用50 mL的乙酸乙酯萃取3次,合并有机相后用饱和的氯化钠水溶液洗涤,再经过无水硫酸钠干燥,所得溶液经过减压浓缩得到粗产品。将粗产品经过柱层析进行分离(V(石油醚)/V(乙酸乙酯)=1∶1),得到白色固体10a。
化合物10a经亨利反应生成化合物11a[9]。1.0 mmol化合物10a和3.0 mmol乙酸铵在硝基甲烷(每1 g化合物10a加20 mL的硝基甲烷)中回流1 h。反应完全后,减压浓缩,加入水和乙酸乙酯形成溶液。用50 mL的乙酸乙酯萃取3次,混合有机相后,用饱和的氯化钠水溶液洗涤,最后用无水硫酸钠干燥,得到粗产品。将所得的粗产品经过柱层析进行分离(V(石油醚)/V(乙酸乙酯)=1∶1),得到黄色固体11a。
化合物11a经氢化铝锂还原反应生成化合物12a[10]。取烘干的圆底烧瓶,加入6 mmol氢化铝锂,氩气保护后,加入10 mL四氢呋喃,在0℃的条件下缓慢滴入体系中。加完原料后,体系逐渐升至室温,在室温下搅拌36 h。反应完全后,用饱和的氢氧化钠水溶液淬灭反应,加入乙酸乙酯。用50 mL的乙酸乙酯萃取3次,合并有机相后用饱和的氯化钠水溶液洗涤,无水硫酸钠干燥,所得溶液经过减压浓缩得到粗产品12a。
化合物12a经酰氯取代反应生成化合物13a。将1.0 mmol化合物12a加入到圆底烧瓶中,氩气保护,加入10 mL二氯甲烷搅拌均匀,再加入1.2 mmol三乙胺。在0℃的条件下,将1.2 mmol氯甲酸甲酯加入到反应体系,均匀搅拌,反应完全后加入适量的水淬灭,用50 mL的二氯甲烷萃取3次,混合有机相后用饱和的氯化钠水溶液洗涤,无水硫酸钠干燥,所得溶液经过减压浓缩得到粗产品。将粗产品经过柱层析进行分离(V(二氯甲烷)/V(甲醇)=50∶1),得到白色固体13a。
化合物13a经三乙基硅烷还原生成化合物14a[11]。将1.0 mmol化合物13a加入到圆底烧瓶中,缓慢加入10 mL三氟乙酸。搅拌均匀后,加入3.0 mmol三乙基硅烷,在60℃的条件下反应12 h。反应完全后,通过减压蒸馏除去大部分的三氟乙酸,然后用饱和的碳酸氢钠水溶液中和剩余的三氟乙酸。随后,用50 mL的二氯甲烷萃取3次,混合有机相后,用饱和的氯化钠水溶液洗涤,无水硫酸钠干燥,得到粗产品。将所得的粗产品经过柱层析进行分离(V(石油醚)/V(乙酸乙酯)=2∶1),得到白色固体14a。
化合物13h经氰基硼氢化钠还原生成化合物14h[12]。将1.0 mmol化合物13h加入到圆底烧瓶中,加入10 mL乙酸搅拌均匀,将体系温度降至0℃搅拌,将10.0 mmol氰基硼氢化钠少量多次的加入到体系中。加完后将体系缓慢地升至室温。反应12 h后,向体系加入二氯甲烷稀释,然后加入1 mol/L的氢氧化钠,将体系的p H增加至大于12。用50 mL的二氯甲烷萃取3次,混合有机相后用饱和的氯化钠水溶液洗涤、无水硫酸钠干燥,得到粗产品。将所得的粗产品经过柱层析进行分离(V(石油醚)/V(乙酸乙酯)=2∶1),得到白色固体14h。
化合物14a经双氧水氧化反应生成化合物15a。将1.0 mmol化合物14a溶于10 mL甲醇中,在0℃的条件下加入0.2 mmol二水合钨酸钠的水溶液,在1 h内缓慢地将10 mmol过氧化氢溶液加入到反应体系中。反应完全后,将反应体系用水洗涤2次后,用饱和的氯化钠水溶液洗涤、无水硫酸钠干燥,得到粗产品。将所得的粗产品经柱层析分离(V(石油醚)/V(乙酸乙酯)=2∶1),得到白色固体15a[2]。
通过上述方法合成了一系列的1⁃羟基色胺衍生物,如图8所示。
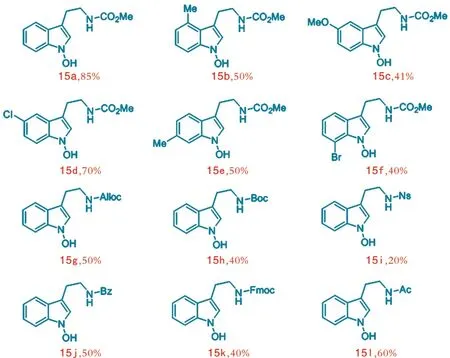
图8 合成的1⁃羟基色胺衍生物
2 结果与讨论
2.1 化合物15l的1 H⁃NMR
图9为化合物15l的1H⁃NMR。1H⁃NMR(500 MHz,甲醇⁃d4)δ:7.53(d,J=8.0 Hz,1H),7.35(d,J=8.2 Hz,1H),7.17~7.12(m,2H),7.05~6.94(m,1H),3.43(t,J=7.3 Hz,2H),2.90(t,J=7.3 Hz,2H),1.90(s,3H)。根据化合物的结构可以推断出7.53和7.35的2个d峰分别是吲哚环上的4⁃位和7⁃位的氢信号,7.17~7.12的2个氢和7.05~6.94的1个氢是吲哚环上5⁃位、6⁃位和2⁃位的氢,3.43和2.90是侧链上的2个亚甲基,1.90是酰基上的甲基。
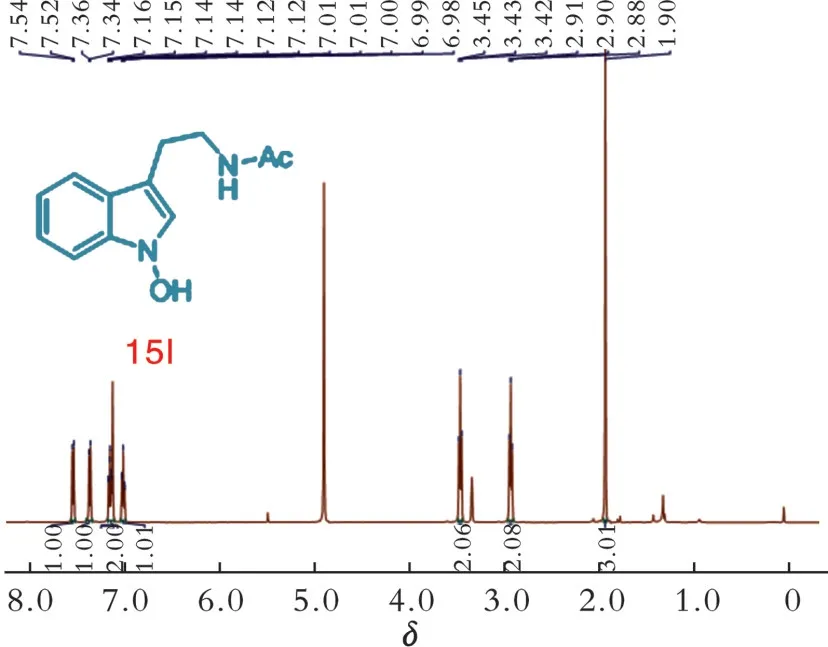
图9 化合物15l的核磁共振氢谱
2.2 化合物15l的13 C⁃NMR
图10为化合物15l的13C⁃NMR。13C⁃NMR(126 MHz,甲醇⁃d4)δ:171.87,134.25,123.67,122.95,121.30,118.30,118.05,107.85,107.39,40.16,24.57,21.21。
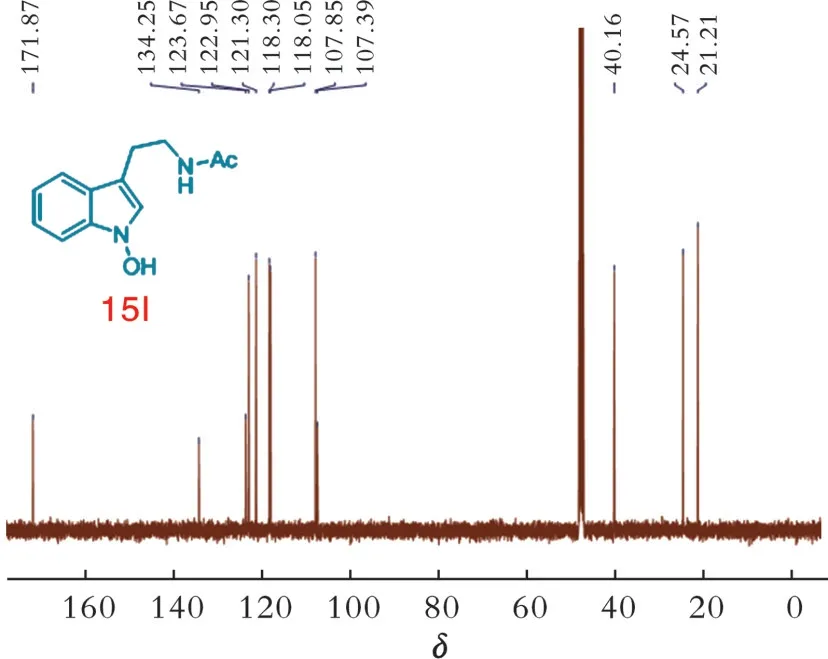
图10 化合物15l的核磁共振碳谱
根据化合物的结构,107.00~134.00都是吲哚环上的碳,共计8个;171.00是酮羰基的碳;21.21~40.16是2个亚甲基以及乙酰基的甲基上的碳,共计3个。
3 结 论
以取代的吲哚为起始底物,经过色胺合成法合成了一系列色胺衍生物。色胺衍生物经过还原、氧化反应得到一系列1⁃羟基色胺衍生物。通过1⁃位羟基优良的亲核性,可以将1⁃羟基色胺衍生物应用于亲核反应、偶联反应以及吲哚生物碱[13⁃16]的合成中。在本方法的基础上,可以继续进行深入的研究,使1⁃羟基色胺衍生物应用于更多方面。
