2,5-二芳基-1,4-苯醌类化合物的合成
2021-03-13杨敏鸽方雨辰朱玉梦朱文庆
杨敏鸽,方雨辰,朱玉梦,张 瑾,樊 盼,朱文庆
(西安工程大学 环境与化学工程学院,陕西 西安 710048)
近年来,苯醌类化合物在生物医药、功能材料、能源、化学合成与分析检测等方面[1-2]的应用日益广泛,许多科研工作者通过对苯醌类化合物进行研究,取得许多成果。部分相关研究工作表明:甲氧基对苯醌具有抗肿瘤的药物活性[3];对苯醌修饰电极,可以提高其工作稳定性及电容量[4-6];2,5-二甲基对苯醌二亚胺作为柔性基团与格式试剂共聚[6]可以制得可溶性苯胺;邻亚甲基苯醌作为一类中间体[7]常用作药物化学及有机合成;具有亲脂性及抗氧化性的艾地苯醌应用于化妆品中可以延缓衰老[8];对二苯醌可以与有机硼烷偶联制备新型芳基化合物[9-10];对苯醌在明胶中以氢键与对苯二酚结合可以改变体系的吸光度[11];对苯醌发生电化学还原反应可以用于浓度测定[12-16]。
虽然苯醌类化合物有着广泛应用,与此同时,我们也发现目前基于苯醌类化合物的合成方法多限于苯醌的烷基化[17-19],关于苯醌的芳基化合成方法鲜有报道。因此,如何实现苯醌芳基化仍是一个亟需解决的科学问题。
本文以对二苯醌为原料通过溴代和Suzuki偶联反应制备出8种2,5-二芳基-1,4-苯醌类化合物4a~4h,收率74%~84%,其结构经1H NMR、13C NMR及HR-MS(ESI-TOF)表征。
为了实现2,5-二芳基-1,4-苯醌类化合物(4)的合成,参考相关文献[20]进行产物合成分析,设计合成反应路线如Scheme 1所示。

Scheme 1
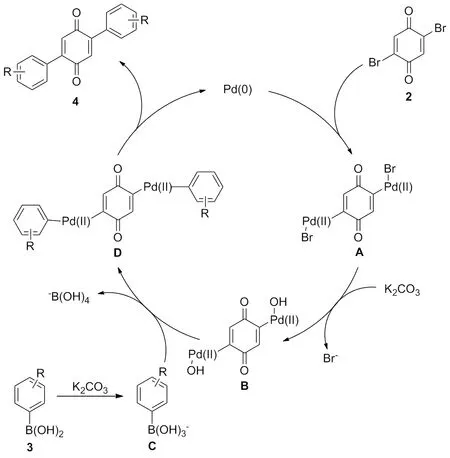
Scheme 2
1 实验部分
1.1 仪器与试剂
X-6型显微熔点仪;Bruker AV 400 MHz型核磁共振仪(TMS为内标);Bruker Esquire 3000plus型高分辨质谱仪;ZF-C型三用紫外分析仪。
所用试剂均为分析纯或化学纯。
1.2 合成
(1) 2,5-二溴-1,4-苯醌(2)的合成[20]
惰性气体保护、冰水浴条件下,称取1,4-苯醌(1)1.05 g(10 mmol)溶解于无水乙醚(45 mL)中,再将提前配制的溴溶液(1.55 g液溴,15 mL三氯甲烷和10 mL无水乙醚)缓慢加入瓶中。室温下反应15 min,缓慢加入25 mL浓硫酸,继续反应35 min。反应结束后,将反应液倒入冰水中并用酸乙酯萃取(3×50 mL),合并有机层,再入氧化银6.0 g(26 mmol),室温反应1 h。反应结束后,经硅胶柱层析 (洗脱剂:石油醚/乙酸乙酯=100/1~20/1,V/V)纯化得2,收率68%。
(2) 化合物4的合成通法
向50 mL圆底烧瓶中依次加入2132.9 mg(0.5 mmol)、芳基硼酸3a~3h(1.25 mmol)、四(三苯基膦)钯(10 mol%)、碳酸钾水溶液(2 mol/L,1.25 mL)和四氢呋喃(5 mL),氩气置换3次,于90 ℃反应6 h,经硅胶柱层析(洗脱剂:石油醚/乙酸乙酯=100/1~20/1,V/V)纯化得8种含有不同芳基取代基的2,5-二芳基-1,4-苯醌类化合物4a~4h,收率74%~84%
4a:黄色固体,收率81%,m.p.215.2~216.6 ℃;1H NMR(400 MHz,CDCl3)δ:7.51~7.59(m,4H),7.42~7.51(m,6H),6.98(s,2H);13C NMR(101 MHz,CDCl3)δ:187.3,145.9,133.5,132.8,130.5,129.7,128.9;HR-MS(ESI-TOF)m/z:Calcd for C18H12O2{[M+H]+}261.0910,found 261.0913。
4b:黄色固体,收率84%,m.p.220.2~221.5 ℃;1H NMR(400 MHz,CDCl3)δ:7.45(d,J=8.1 Hz,4H),7.26(d,J=9.0 Hz,4H),6.92(s,2H),2.40(s,6H);13C NMR(101 MHz,CDCl3)δ:187.6,145.8,140.8,132.8,130.0,129.6,129.6,21.7;HR-MS(ESI-TOF)m/z:Calcd for C20H16O2{[M+H]+}289.1223,found 289.1230。
4c:黄色固体,收率78%,m.p.225.1~226.4 ℃;1H NMR(400 MHz,CDCl3)δ:7.49(d,J=7.9 Hz,4H),7.31(d,J=7.9 Hz,4H),6.95(s,2H),2.71(q,J=7.6 Hz,4H),1.28(t,J=7.6 Hz,6H);13C NMR(101 MHz,CDCl3)δ:187.6,147.0,145.7,132.8,130.2,129.7,128.5,29.1,15.7;HR-MS(ESI-TOF)m/z:Calcd for C22H20O2{[M+H]+}317.1536,found 317.1539。
4d:黄色固体,收率74%,m.p.233.0~234.0 ℃;1H NMR(400 MHz,CDCl3)δ:7.48(d,J=7.7 Hz,4H),7.27(d,J=7.7 Hz,4H),6.94(s,2H),2.64(t,J=7.5 Hz,4H),1.80~1.62(m,4H),0.97(t,J=7.2 Hz,6H);13C NMR(101 MHz,CDCl3)δ:187.6,145.7,145.5,132.8,130.2,129.6,129.1,38.2,24.7,14.2;HR-MS(ESI-TOF)m/z:Calcd for C24H24O2{[M+H]+}345.1849,found 345.1849。
4e:黄色固体,收率80%,m.p.223.1~225.4 ℃;1H NMR(400 MHz,CDCl3)δ:7.50(d,J=8.1 Hz,4H),7.33(d,J=8.1 Hz,4H),6.95(s,2H),2.97(m,2H),1.29(d,J=6.9 Hz,12H);13C NMR(101 MHz,CDCl3)δ:187.6,151.6,145.8,132.9,130.3,129.7,127.1,34.4,24.2;HR-MS(ESI-TOF)m/z:Calcd for C24H24O2{[M+H]+}345.1849,found 345.1851。
4f:黄色固体,收率79%,m.p.226.5~227.8 ℃;1H NMR(400 MHz,CDCl3)δ:7.47(d,J=21.2 Hz,8H),6.96(s,2H),1.36(s,18H);13C NMR(101 MHz,CDCl3)δ:187.6,153.9,145.7,132.9,130.0,129.5,126.0,35.2,31.5;HR-MS(ESI-TOF)m/z:Calcd for C26H28O2{[M+H]+}373.2162,found 373.2168。
4g:黄色固体,收率79%,m.p.210.4~211.0 ℃;1H NMR(400 MHz,CDCl3)δ:7.31~7.35(m,8H),6.95(s,2H),2.43(s,6H);13C NMR(101 MHz,CDCl3)δ:187.4,146.1,138.6,133.4,132.8,131.2,130.2,128.8,126.8,21.8;HR-MS(ESI-TOF)m/z:Calcd for C20H16O2{[M+H]+}289.1223,found 289.1230。
4h:黄色固体,收率76%,m.p.223.6~224.2 ℃;1H NMR(400 MHz,CDCl3)δ:7.50(d,J=8.6 Hz,4H),7.44(d,J=8.6 Hz,4H),6.95(s,2H);13C NMR(101 MHz,CDCl3)δ:186.8,144.9,137.1,133.4,131.0,130.9,129.3;HR-MS(ESI-TOF)m/z:Calcd for C18H10Cl2O2{[M+H]+}329.0131,found 329.0131。
2 结果与讨论
(1) 条件优化
以2与3a的Suzuki偶联反应为模板,进一步筛选和优化了催化剂、温度、溶剂、无机碱等反应条件。
发生Suzuki偶联反应过程中需要零价钯参与。含氧原子和氮原子本身具有弱的配位能力,能够阻止零价钯的聚集,保持金属钯的催化活性。基于此,首先探究了甲苯(Toluene)、异丙醇(IPA)、N,N-二甲基甲酰胺(DMF)、四氢呋喃(THF)这四种实验室常用的有机溶剂对模板反应的影响。如表1所示,反应收率为34%~56%,其中以四氢呋喃(THF)为最适溶剂(Entry 4),产率达到56%。保持其它条件不变,在最适溶剂为四氢呋喃(THF)的条件下,进一步考察了碱对反应的影响。以K2CO3为无机碱时,模板反应的产率最高(Entry 6),达到62%。随后筛选了碱的用量,结果表明,2.5 eq.碱为最佳用量(Entry 6),产率提升到68%。在最适溶剂为四氢呋喃(THF)及最适碱为K2CO3(2.5 eq.)条件下,保持其它条件不变,对实验所需的不同钯催化剂进行考察,其它钯催化条件下并没有提升产率(Entry 6),最适催化剂仍为Pd(PPh3)4。随后进行了钯催化剂用量的筛选,对比实验结果发现钯催化剂用量为10 mol%最佳(Entry 6),产率达到76%。在确定最适溶剂、碱、催化剂及当量条件下,通过进一步筛选反应温度,确定90 ℃反应为最优,产率达到81%。最后通过延长或缩短反应时间以及改变硼酸用量,产率均没有更大提升。
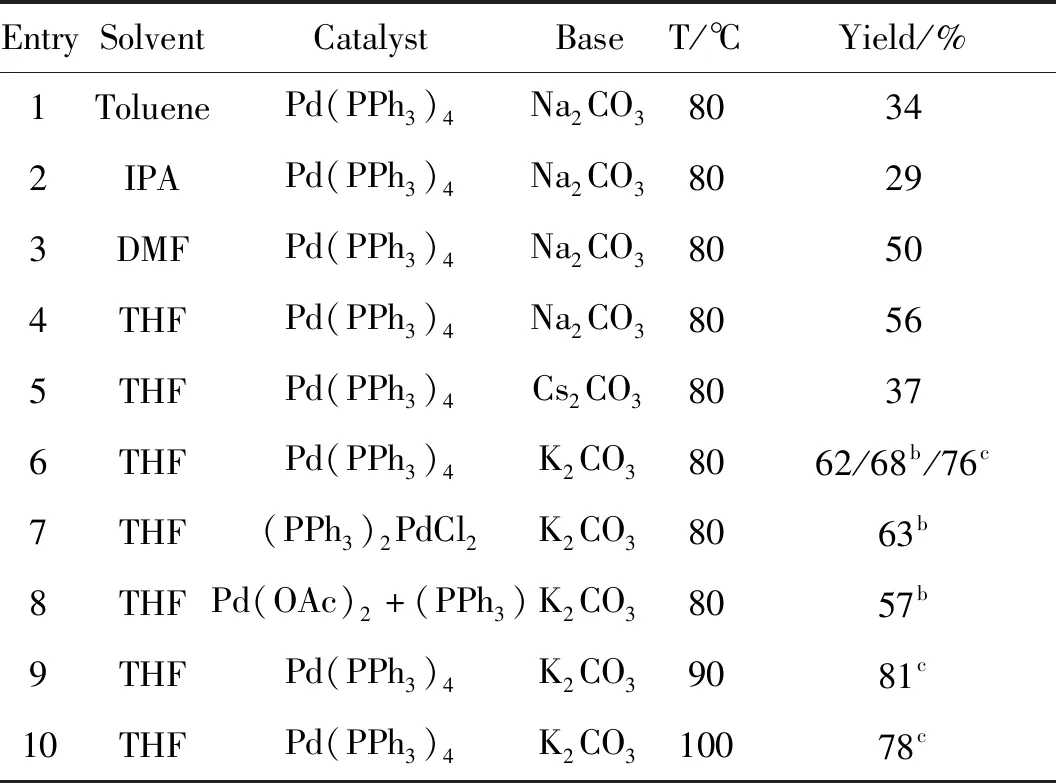
表1 反应条件的筛选与优化Table 1 Screening and optimization of reaction conditions a
(2) 目标产物
发生Suzuki偶联反应过程中,硼酸上所连芳基取代基的电子效应差异使得目标产物的产率各不相同。对比目标产物的收率可知,当硼酸的芳基上带有甲基、乙基供等电子基时,偶联反应产率较高,如产物4b、4c和4e;而芳基连有吸电子基团时,产率偏低,如产物4h。对比4b和4g可以看出,对位取代产物的产率优于间位。取代基的位点不同进一步分析可以看出,受到取代基空间位阻的效应的影响,位阻增大,目标产物的反应产率也略有下降。
2,5-二溴-1,4-苯醌(2)首先与Pd(0)氧化加成,生成溴代芳烃中间体A,随后再与碳酸钾反应脱溴生成含有强极性键(Pd—O)的中间体B;另一分子的碳酸钾将芳基硼酸3活化,生成富电性较强的硼酸盐中间体C,使阴离子更利于向中间体B的金属中心迁移。中间体B与中间体C通过转金属过程生成有机钯络合物D,最后经还原消除生成目标产物4和Pd(0)。接着,Pd(0)进入下一个催化循环[21]。
利用Suzuki偶联反应合成了8种新型含芳基2,5-二芳基-1,4-苯醌类化合物。目标产物中带有给电子取代基产物的产率比吸电子取代基产物的产率高;进一步对比产率可知,对位取代优于间位;目标产物产率受芳基上取代基空间位阻影响,取代基空间位阻增大,反应产率降低。该合成方法为制备新型含芳基的二芳基苯醌类化合物提供了一条更加便捷高效的途径。
