新型苯并三噻吩衍生物的合成及性能
2021-03-13吴春林陈学刚
唐 涛,吴春林,陈学刚
(青岛科技大学 高分子科学与工程学院,山东 青岛 266000)
噻吩及其衍生物,如联噻吩、并二噻吩、并三噻吩、苯并二噻吩(BDT)等,具有优异的供电性及载流子传输特性,经常用于共轭聚合物及电子给-受体(D-A)体系的构筑[1],并将其应用于有机光伏(OPV)电池材料及器件[2-5],有机发光二极管(OLEDs)[6-7],有机场效应晶体管(OFETs)[8-11]和电致变色显示[12-13]等领域中。与苯并二噻吩相比,近年出现的苯并三噻吩(BTT)不仅保持较强的供电子特性,而且具有比BDT更为扩展的共轭平面结构,因此非常有利于固态的π-π*堆积及载流子的高效传输,成为可以与BDT比肩的一类具有应用前景的新型供电性单元,在D-A共聚物的设计及新型光电材料领域的研究中引起重视[14-15]。
本文以2,3-二溴噻吩(1)为原料,制得BTT(6);并在此基础上合成BTT有机锡中间体(7),然后与2-溴-3-正十二烷基噻吩发生Stille偶联反应,对其共轭程度进行扩展,合成了2,8-二(3-正十二烷基噻吩)-5-(1-辛基壬基)苯并[2,1-b:3,4-b′:5,6-b″]三噻吩(8,BTT-T,Scheme 1)。

Scheme1
1 实验部分
1.1 仪器与试剂
Bruker Avance 500 MHz型超导核磁共振仪(TMS为内标,CDCl3为溶剂);Hitachi U-4100型紫外可见近红外分光光度计;Hitachi F-4600型荧光光谱仪;Autolab PGSTAT 204型电化学工作站;Bruker Impact II型四极杆飞行时间质谱仪;NETZSCH TG 209 F1型热重分析仪。
所用试剂均为分析纯。
1.2 合成
(1) 2,3-二溴-5-壬酰基噻吩(2)的合成
将2,3-二溴噻吩1.26 g(5.21 mmol)溶于10 mL干燥二氯甲烷中,冰水浴冷却,氮气保护下滴加壬酰氯1.12 mL(5.85 mmol),滴毕,搅拌10 min;加入无水三氯化铝0.92 g(6.85 mmol),于0℃反应2 h。用2 M盐酸20 mL淬灭反应,二氯甲烷萃取,无水Na2SO4干燥,过滤,滤液旋蒸除溶,残余物真空干燥得黄棕色油状液体21.89 g,产率95.1%;1H NMRδ:7.47(s,1H),2.80(t,J=6.0 Hz,2H),1.74~1.69(m,2H),1.36~1.26(m,10H),0.88(t,J=5.6 Hz,3H)。
(2) 5-壬酰基-2,3-二(3-噻吩)噻吩(3)的合成
将化合物21.34 g(3.50 mmol),噻吩-3-硼酸1.05 g(8.20 mmol),碳酸钠4.19 g(40.02 mmol)溶于混合溶剂中(11 mL甲苯,11 mL乙醇,11 mL去离子水),氮气保护下,加入四三苯基膦钯0.11 g(0.10 mmol),回流反应24 h。用水淬灭反应,乙醚萃取,无水Na2SO4干燥,过滤,滤液旋蒸除溶,粗产物经硅胶柱层析(洗脱剂:A=石油醚/乙酸乙酯=8/1,V/V)纯化,真空干燥得黄色固体31.07 g,产率79.5%;1H NMRδ:7.70(s,1H),7.34(d,J=3.8 Hz,1H),7.28(m,2H),7.25(m,1H),7.01(m,2H),2.90(t,J=7.5 Hz,2H),1.78(m,2H),1.29~1.26(m,10H),0.90(t,J=6.5 Hz,3H)。
(3) 5-壬酰基苯并[2,1-b:3,4-b′:5,6-b″]三噻吩(4)的合成
将化合物30.60 g(1.54 mmol)溶于40 mL干燥二氯甲烷中,冰水浴冷却,氮气保护下加入三氟化硼乙醚0.31 mL(2.47 mmol)和2,3-二氯-5,6-二氰基-1,4-苯醌0.53 g(2.35 mmol),反应48 h。加入锌粉0.91 g(13.90 mmol)和20 mL甲醇淬灭反应。滤除不溶物,用水洗涤,无水Na2SO4干燥,过滤,滤液旋蒸除溶,粗产物经硅胶柱层析(洗脱剂:A=10/1)纯化,真空干燥得橘黄色固体40.24 g,产率39.9%;1H NMRδ:8.31(s,1H),7.76(d,J=5.5 Hz,1H),7.64(d,J=5.5 Hz,1H),7.55(d,J=5.5 Hz,1H),7.53(d,J=5.5 Hz,1H),3.08(t,J=7.0 Hz,2H),1.85(m,2H),1.48~1.31(m,10H),0.90(t,J=7.0 Hz,3H)。
(4) 5-((1-羟基-1-辛基)壬基)苯并[2,1-b:3,4-b′:5,6-b″]三噻吩(5)的合成
将化合物40.51 g(1.29 mmol)溶于25 mL干燥乙醚中,冰水浴冷却,氮气保护下滴加2 M辛基溴化镁2.19 mmol的乙醚(1.10 mL)溶液,于0 ℃搅拌反应1 h;回流反应过夜。冷却至室温,用饱和氯化铵溶液18 mL淬灭反应,乙醚萃取,无水Na2SO4干燥,过滤,滤液旋蒸除溶,粗产物经硅胶柱层析 (洗脱剂:A=15/1)纯化,真空干燥得淡黄色固体50.61 g,产率93.5%;1H NMRδ:7.73(d,J=5.5 Hz,1H),7.57(d,J=5.0 Hz,1H),7.52(s,1H),7.50(d,J=4.0 Hz,1H),7.49(d,J=4.0 Hz,1H),2.14(s,1H),1.94(m,4H),1.45~1.37(m,4H),1.25~1.21(m,20H),0.84(t,J=6.5 Hz,6H)。
(5) 5-(1-辛基壬基)苯并[2,1-b:3,4-b′:5,6-b″]三噻吩(6)的合成
将氢化铝锂0.40 g(10.47 mmol)加入反应瓶中,冰水浴冷却,缓慢滴加无水乙醚30 mL,滴毕,搅拌使其溶解;氮气保护下,分批加入无水三氯化铝0.54 g(4.03 mmol),加毕,于0 ℃搅拌30 min;滴加化合物50.48 g(0.95 mmol)的无水乙醚(10 mL)溶液,滴毕,于室温反应36 h。用冰水中和氢化铝锂,乙醚萃取,无水Na2SO4干燥,过滤,滤液旋蒸除溶,粗产物经硅胶柱层析(洗脱剂:石油醚)纯化,真空干燥得白色固体60.41 g,产率83.9%;1H NMRδ:7.70(d,J=5.0 Hz,1H),7.56(d,J=5.0 Hz,1H),7.47(d,J=5.0 Hz,1H),7.46(d,J=5.0 Hz,1H),7.40(s,1H),2.97(m,1H),1.79~1.65(m,4H),1.28~1.22(m,24H),0.84(t,J=6.5 Hz,6H);13C NMRδ:150.38,132.58,132.38,131.67,131.51,130.80,129.71,124.82,124.06,122.84,122.31,119.17,42.43,38.06,31.88,29.66,29.51,29.32,27.57,22.66,14.12;HR-MS(ESI)m/z:Calcd for C29H41S3{[M+H]+}485.24,found 485.26。
(6) 2,8-二(三甲基锡)-5-(1-辛基壬基)苯并[2,1-b:3,4-b′:5,6-b″]三噻吩(7)的合成
将化合物60.30 g(0.62 mmol)溶于干燥四氢呋喃中,氮气保护下,于-78 ℃搅拌10 min;滴加1.6 M正丁基锂(2.46 mmol)的己烷(1.55 mL)溶液,滴毕,反应1 h;于室温反应1 h;于-78 ℃反应10 min。滴加1 M三甲基氯化锡(2.46 mmol)的己烷(2.45 mL)溶液,滴毕,反应1 h;于室温反应过夜。加水淬灭反应,用二氯甲烷萃取,无水Na2SO4干燥,过滤,滤液旋蒸除溶,残余物真空干燥得淡黄色油状液体70.36 g,产率73.0%;1H NMRδ:7.77(s,1H),7.61(s,1H),7.45(s,1H),2.97(m,1H),1.77~1.65(m,4H),1.24~1.21(m,24H),0.85(t,J=7.0 Hz,6H),0.46(t,J=3.5 Hz,18H)。
(7) 2,8-二(3-正十二烷基噻吩)-5-(1-辛基壬基)苯并[2,1-b:3,4-b′:5,6-b″]三噻吩(8)的合成
将化合物70.20 g(0.25mmol)和2-溴-3-正十二烷基噻吩0.18 g(0.54 mmol)溶于15 mL甲苯中,氮气保护下加入四三苯基磷钯0.01 g(0.001 mmol),于110 ℃回流反应24 h。乙酸乙酯萃取,无水Na2SO4干燥,过滤,滤液旋蒸除溶,粗产物经硅胶柱层析(洗脱剂:正己烷)纯化,真空干燥得黄色固体80.14 g,产率56.8%;1H NMRδ:7.68(s,1H),7.53(s,1H),7.39(s,1H),7.27(d,J=5.0 Hz,2H),7.00(d,J=4.8 Hz,2H),2.99(dd,J=9.3 Hz,4.7 Hz,1H),2.90(dd,J=14.8 Hz,6.2 Hz,4H),1.73(m,8H),1.45~1.21(m,60H),0.85(q,J=7.0 Hz,12H);13C NMRδ:150.66,140.83,140.67,135.51,134.70,132.54,131.89,130.65,130.58,130.28,128.98,124.75,124.59,121.25,120.44,119.17,42.50,38.08,31.93,30.72,29.68,29.51,29.34,27.60,22.69,14.13;HR-MS(ESI)m/z:Calcd for C61H93S5{[M+H]+}986.58,found 986.39。
2 结果与讨论
2.1 光物理性质
图1为两种分子的UV-Vis谱图。由图1可知,在溶液态(三氯甲烷)中,BTT的吸收范围为240~370 nm,BTT-T的吸收范围为240~460 nm。这正是因为BTT-T分子两侧引入了噻吩单元所致。两侧噻吩单元的引入,使其共轭长度显著增加,导致吸收范围扩大。BTT分子在278 nm处的吸收峰和310 nm处的肩峰,是由π-π*跃迁产生的;与BTT相比,BTT-T除了 293 nm处的π-π*跃迁机制产生的吸收峰之外,还在372 nm附近产生较强的吸收带,这是因为两侧强供电性噻吩基团的引入,导致分子在被激发时,同时产生了分子内电荷转移跃迁机制。
在固体状态下,BTT和BTT-T的UV-Vis谱图与溶液态谱图大致相似,不同之处在于其吸收峰整体出现少许红移,且峰形变宽。这是因为固态下分子之间发生聚集导致的,符合大多数共轭分子的一般特征。通过其固态UV-Vis谱图,计算得到BTT和BTT-T的光学能隙分别为3.44 eV和2.75 eV。由此可见,BTT-T由于两侧引入的噻吩基团,使其共轭长度增加,导致能隙显著降低。
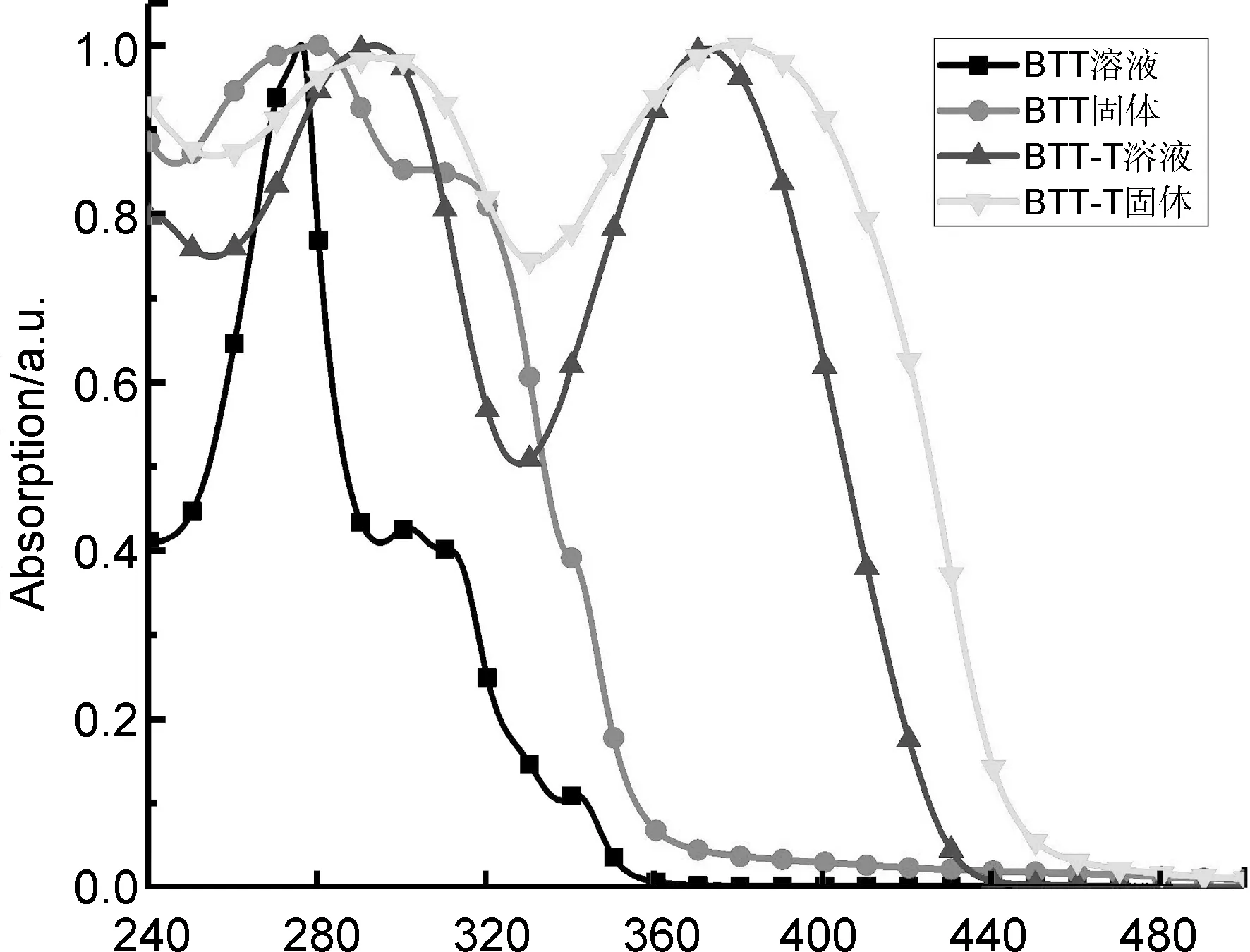
λ/nm图1 BTT和BTT-T的UV-Vis谱图Figure 1The UV-Vis spectra of BTT and BTT-T

λ/nm图2 BTT和BTT-T的FL谱图Figure 2The FL spectra of BTT and BTT-T
图2为两种分子在溶液中(三氯甲烷)及固态时的FL谱图(激发波长300 nm)。由图3可知,在溶液态中,BTT及BTT-T分别在372 nm及461 nm附近有荧光发射峰。由于在BTT-T中,噻吩基团的引入导致其共轭程度增加,吸收及发射的能量降低,发射峰出现较大程度的红移(89nm)。在固态中,BTT及BTT-T的荧光发射峰分别位于380 nm及488 nm。与溶液态相比,表现出一定程度的红移,与UV-Vis谱图一致,这也是由于固体状态下,分子间聚集引发的。
2.2 电化学学性质
图3为BTT和BTT-T的CV曲线。由图3可知,BTT和BTT-T的起始氧化电位分别为0.8 eV和0.75 eV,估算得到BTT和BTTT-T的HOMO能级分别为-5.14 eV和-5.09 eV。较高的HOMO能级,说明BTT-T的氧化性比BTT要强。BTT-T中引入了强给电子的噻吩基团,共轭程度和给电子能力都显著增强,导致其在正电位扫描的过程中,更易被氧化。同时,在BTT-T的曲线里还出现了第二个氧化峰,说明BTT-T可以连续失电子,呈现出多种氧化状态。在两种分子中,基本都由强给电性的噻吩单元构成,吸电子能力弱,分子难以被还原,因此测定未获得明显的还原电位,在负电位扫描中表现出较强的稳定性。

Voltage/V图3 BTT和BTT-T的循环伏安曲线Figure 3The cyclic voltammograms of BTT and BTT-T

Temperature/℃图 4 BTT和BTT-T的TGA曲线Figure 4The TGA curves of BTT and BTT-T
2.3 热稳定性
图4为BTT和BTT-T的TGA曲线。由图4可知,BTT和BTT-T均表现出较好的热稳定性。与BTT相比,BTT-T的热稳定性更优。这是因为BTT-T的分子体系中有更长的共轭体系,使得键能增强,热稳定性增加。
通过Stille偶联法,成功合成了共轭长度扩展的苯并三噻吩衍生物BTT-T。强供电性基团噻吩的引入,显著提高了分子的共轭程度,导致其紫外-可见吸收范围明显扩展,呈现出窄带隙特征,有望作为高性能光电材料加以应用。
