2-硝基苯并呋喃与邻巯基苯甲醛反应:一锅法合成硫代黄酮衍生物
2021-03-13卓俊睿赵建强袁伟成1
卓俊睿,赵建强,袁伟成1,
(1.中国科学院 成都有机化学研究所,四川 成都 610041;2.成都大学 高等研究院,四川 成都 6101062;3.中国科学院大学,北京 100049)
硫代色满酮衍生物是一类重要的结构单元,常见于天然产物及活性分子中(Chart 1)[1],具有抗炎、抗癌和抗菌等活性[2-4]。如化合物A表现出体外的人类凝固酵素或者凝血酶抑制作用[5];化合物B可以预防和治疗类固醇硫酸酯酶缺乏所致的疾病[6];化合物C具有除草生物活性,可以用作除草剂或者除草剂的药物中间体[7];化合物D具有加速尿酸排泄及抑制黄嘌呤氧化酶的生物活性[8];化合物E可治疗肾上腺素失调引起的病症[9],而化合物F具有体外抑制肾上腺素活性的药理作用[10]。鉴于其广泛的生理活性,研究人员已开发了诸多合成硫代色满酮衍生物的方法。例如含有硫代色烯骨架的分子发生分子内Friedel Craft-氧化级联反应[11],过渡金属催化硫代色酮骨架的分子内环化[12-13],芳基巯基试剂与硫代色酮的sulfur-Michael/aldol反应等[14]。虽然目前已报道的方法已为该类化合物的合成提供了可行的方案,但仍普遍存在一些难以避免的缺点:部分分子内环化反应涉及金属催化剂[15-16];步骤繁琐,前期底物即需要多步反应才能获得[17];反应需要较为苛刻的条件,对产物官能团耐受性提出挑战[18]。

Chart 1
本文通过一锅三步反应实现硫代色满酮衍生物的合成。2-硝基苯并呋喃(1a~1k)和邻巯基苯甲醛(2)在甲苯中被三乙烯二胺(DABCO)拔氢得到去芳构化的加成产物,随后在二氮杂二环(DBU)的作用下,生成脱除亚硝酸的中间体;中间体极不稳定,易被空气中的氧气氧化为终产物3a~3k(Scheme 1),收率42%~67%,其结构经1H NMR,13C NMR及HR-MS(ESI-TOF)确证。
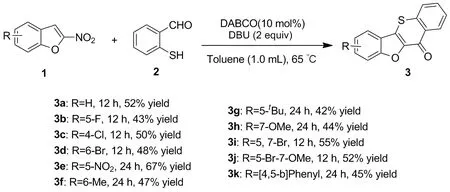
Scheme 1
1 实验部分
1.1 仪器与试剂
BüchiB-545型熔点仪;ZF-2型三用紫外分析仪;Brucker-300 MHz型核磁共振仪(TMS为内标);Bruker Q TOF型高分辨质谱质谱仪。
所用试剂均为分析纯。
1.2 化合物3a~3k的合成(以3a为例)
向干燥反应管中加入2-硝基苯并呋喃(1a)16.3 mg(0.1 mmol)和三乙烯二胺1 mg(0.01 mmol),甲苯(1 mL)为溶剂,依次加入邻巯基苯甲醛216.5 mg(0.12 mmol)和DBU 30.6 mg(0.2 mmol),搅拌下于65 ℃反应12 h(TLC检测)。冷却至室温,旋蒸除溶,用少量二氯甲烷溶解粗产物,经硅胶柱层析[洗脱剂:V(乙酸乙酯)/V(石油醚)=1/10~1/5]纯化得白色固体3a13.1 mg,收率52%。
用类似的方法合成化合物3b~3k。
11H-硫代色满酮[3,2-b]苯并呋喃-11-酮(3a):白色固体,收率52%,m.p.219~220 ℃;1H NMR(300 MHz,CDCl3)δ:8.75(dd,J=8.1 Hz,1.4 Hz,1H),7.80(d,J=7.9 Hz,1H),7.78~7.70(m,2H),7.69~7.54(m,3H),7.45~7.38(m,1H);13C NMR(75 MHz,CDCl3)δ:170.1,155.2,144.6,135.6,133.2,131.4,130.1,129.2,127.0,126.8,125.5,124.4,124.0,121.2,113.2;HR-MS(ESI-TOF)m/z:Calcd for C15H8O2SNa{[M+Na]+}275.0137,found 275.0136。
7-氟-11H-硫代色满酮[3,2-b]苯并呋喃-11-酮(3b):白色固体,收率43%,m.p.289.7~290.5 ℃;1H NMR(300 MHz,CDCl3)δ:8.77(dd,J=8.0 Hz,1.6 Hz,1H),7.80(d,J=7.4 Hz,1H),7.75~7.66(m,2H),7.66~7.58(m,1H),7.49(dd,J=7.7 Hz,2.6 Hz,1H),7.41~7.32(m,1H);13C NMR(75 MHz,CDCl3)δ:170.2,159.4(J=241.5 Hz),151.5,145.9,135.5,133.1,131.7,129.3,127.1,127.0,125.2,125.0,118.3(J=26.3 Hz),114.4(J=9.0 Hz),106.8(J=25.5 Hz);HR-MS(ESI-TOF)m/z:Calcd.for C15H7O2SFNa{[M+Na]+}293.0043,found 293.0035。
6-氯-11H-硫代色满酮[3,2-b]苯并呋喃-11-酮(3c):白色固体,收率50%,m.p.272.4~272.9 ℃;1H NMR(600 MHz,CDCl3)δ:8.81~8.75(m,1H),7.82(d,J=8.1 Hz,1H),7.75~7.69(m,1H),7.68~7.60(m,2H),7.55(t,J=8.1 Hz,1H),7.41(d,J=7.8 Hz,1H);13C NMR(151 MHz,CDCl3)δ:170.5,156.1,145.3,136.9,133.2,131.9,130.7,129.3,128.7,127.5,127.4,125.5,124.6,123.9,112.1;HR-MS(ESI-TOF)m/z:Calcd for C15H7O2SClNa{[M+Na]+}308.9747,found 308.9746。
8-溴-11H-硫代色满酮[3,2-b]苯并呋喃-11-酮(3d):白色固体,收率48%,m.p.276.8~277.3 ℃;1H NMR(300 MHz,CDCl3)δ:8.77(dd,J=7.9 Hz,1.3 Hz,1H),7.92(d,J=1.2 Hz,1H),7.79(d,J=8.1 Hz,1H),7.75~7.66(m,2H),7.66~7.54(m,2H);13C NMR(75 MHz,CDCl3)δ:170.1,155.4,144.9,137.1,135.4,133.2,131.6,129.3,127.8,127.1,125.3,123.9,123.5,122.1,116.7;HR-MS(ESI-TOF)m/z:Calcd for C15H7O2SBrNa{[M+Na]+}352.9242,found 352.9248。
7-硝基-11H-硫代色满酮[3,2-b]苯并呋喃-11-酮(3e):黄色固体,收率67%,m.p.297.3~297.8 ℃;1H NMR(600 MHz,CDCl3)δ:8.86~8.74(m,2H),8.55(dd,J=9.2 Hz,2.3 Hz,1H),7.92~7.82(m,2H),7.78~7.73(m,1H),7.67(t,J=7.5 Hz,1H);13C NMR(151 MHz,CDCl3)δ:170.4,158.1,147.1,145.0,135.6,133.5,132.5,129.8,127.9,127.65,126.0,125.5,125.4,118.5,114.3 ;HR-MS(ESI-TOF)m/z:Calcd for C15H7NO4SNa{[M+Na]+}319.9988,found 319.9993。
8-甲基-11H-硫代色满酮[3,2-b]苯并呋喃-11-酮(3f):白色固体,收率47%,m.p.269.7~270.3 ℃;1H NMR(300 MHz,CDCl3)δ:8.76(d,J=7.9 Hz,1H),7.76(d,J=7.5 Hz,1H),7.72~7.62(m,2H),7.62~7.54(m,1H),7.52(s,1H),7.25(d,J=3.5 Hz,1H),2.55(s,3H);13C NMR(75 MHz,CDCl3)δ:170.0,155.8,144.3,141.4,135.6,133.2,131.2,129.1,127.0,126.8,125.8,125.7,121.9,120.6,113.1,22.3;HR-MS(ESI-TOF)m/z:Calcd for C16H10O2SNa{[M+Na]+}289.0294,found 289.0296。
7-叔丁基-11H-硫代色满酮[3,2-b]苯并呋喃-11-酮(3g):白色固体,收率42%,m.p.176.8~177.2 ℃;1H NMR(300 MHz,CDCl3)δ:8.83~8.72(m,1H),7.84~7.73(m,2H),7.73~7.62(m,3H),7.62~7.54(m,1H),1.43(s,9H);13C NMR(75 MHz,CDCl3)δ:170.1,153.6,147.5,144.8,135.6,133.2,131.3,129.1,128.6,127.0,126.8,125.8,124.1,116.9,112.6,35.0,31.6;HR-MS(ESI-TOF)m/z:Calcd for C19H16O2SNa{[M+Na]+}331.0763,found 331.0762。
9-甲氧基-11H-硫代色满酮[3,2-b]苯并呋喃-11-酮(3h):白色固体,收率44%,m.p.208.8~209.8 ℃;1H NMR(300 MHz,CDCl3)δ:8.73(dd,J=8.1 Hz,1.3 Hz,1H),7.75~7.68(m,1H),7.66~7.58(m,1H),7.57~7.50(m,1H),7.32~7.26(m,2H),7.03(dd,J=6.6 Hz,2.4 Hz,1H),4.04(s,3H);13C NMR(75 MHz,CDCl3)δ:169.8,146.4,145.0,144.5,135.5,133.2,131.2,129.1,126.9,126.7,125.8,125.5,124.8,112.5,111.5,56.3;HR-MS(ESI-TOF)m/z:Calcd for C22H19N2O6Na{[M+Na]+}407.1238,found 407.1228。
7,9-二溴-11H-硫代色满酮[3,2-b]苯并呋喃-11-酮(3i):白色固体,收率55%,m.p.277.0~277.9 ℃;1H NMR(300 MHz,CDCl3)δ:8.777.75~7.68(m,1H),7.66~7.59(m,1H);13C NMR(75 MHz,CDCl3)δ:169.8,151.7,145.5,135.2,133.2,131.9,129.4,127.3,127.1,126.9,124.6,122.8,117.2,107.0;HR-MS(ESI-TOF)m/z:Calcd for C15H6O2SBr2Na{[M+Na]+}430.8347,found 430.8350。
7-溴-9-甲氧基-11H-硫代色满酮[3,2-b]苯并呋喃-11-酮(3j):白色固体,收率52%,m.p.279.8~280.2 ℃;1H NMR(300 MHz,CDCl3)δ:8.77(d,J=8.0 Hz,1H),7.78(d,J=8.0 Hz,1H),7.69(t,J=7.5 Hz,1H),7.61(t,J=7.3 Hz,1H),7.53(d,J=1.6 Hz,1H),7.18(d,J=1.4 Hz,1H),4.07(s,3H);13C NMR(151 MHz,CDCl3+CD3OD)δ:170.5,147.2,145.4,144.5,136.0,133.3,132.1,129.5,127.5,127.3,125.5,117.7,115.6,115.5,56.9;HR-MS(ESI-TOF)m/z:Calcd for C16H9BrO3SNa{[M+Na]+}382.9348,found 382.9348。
8H-萘[2,1-b]硫代色满酮[2,3-d]呋喃-8-酮(3k):白色固体,收率45%,m.p.283.3~283.8 ℃;1H NMR(300 MHz,CDCl3)δ:8.79(d,J=8.1 Hz,1H),8.30(d,J=8.3 Hz,1H),8.01(d,J=8.7 Hz,2H),7.82(d,J=7.5 Hz,2H),7.78~7.67(m,2H),7.65~7.56(m,2H);13C NMR(75 MHz,CDCl3)δ:169.5,154.4,136.0,132.9,131.9,131.4,130.6,129.5,129.1,128.2,127.8,126.9,126.9,126.0,125.7,123.4,118.8,113.1;HR-MS(ESI-TOF)m/z:Calcd for C19H10O2SNa{[M+Na]+}325.0294,found 325.0300。
2 结果与讨论
2.1 反应条件优化
该反应合成硫代黄酮衍生物的历程分为3步:第一步为去芳香化[4+2]环加成反应,该过程可以通过邻巯基苯甲醛对2-硝基苯并呋喃的亲核进攻实现;第二步为碱介导的亚硝酸的消除反应;第三步为空气中的氧气与脱除亚硝酸后不稳定的中间体发生的氧化反应。最终三步反应通过一锅法实现,中间无需进行中间体的分离。以2-硝基苯并呋喃1a和邻巯基苯甲醛2的反应为模板,对反应条件进行筛选,实验结果见表1。
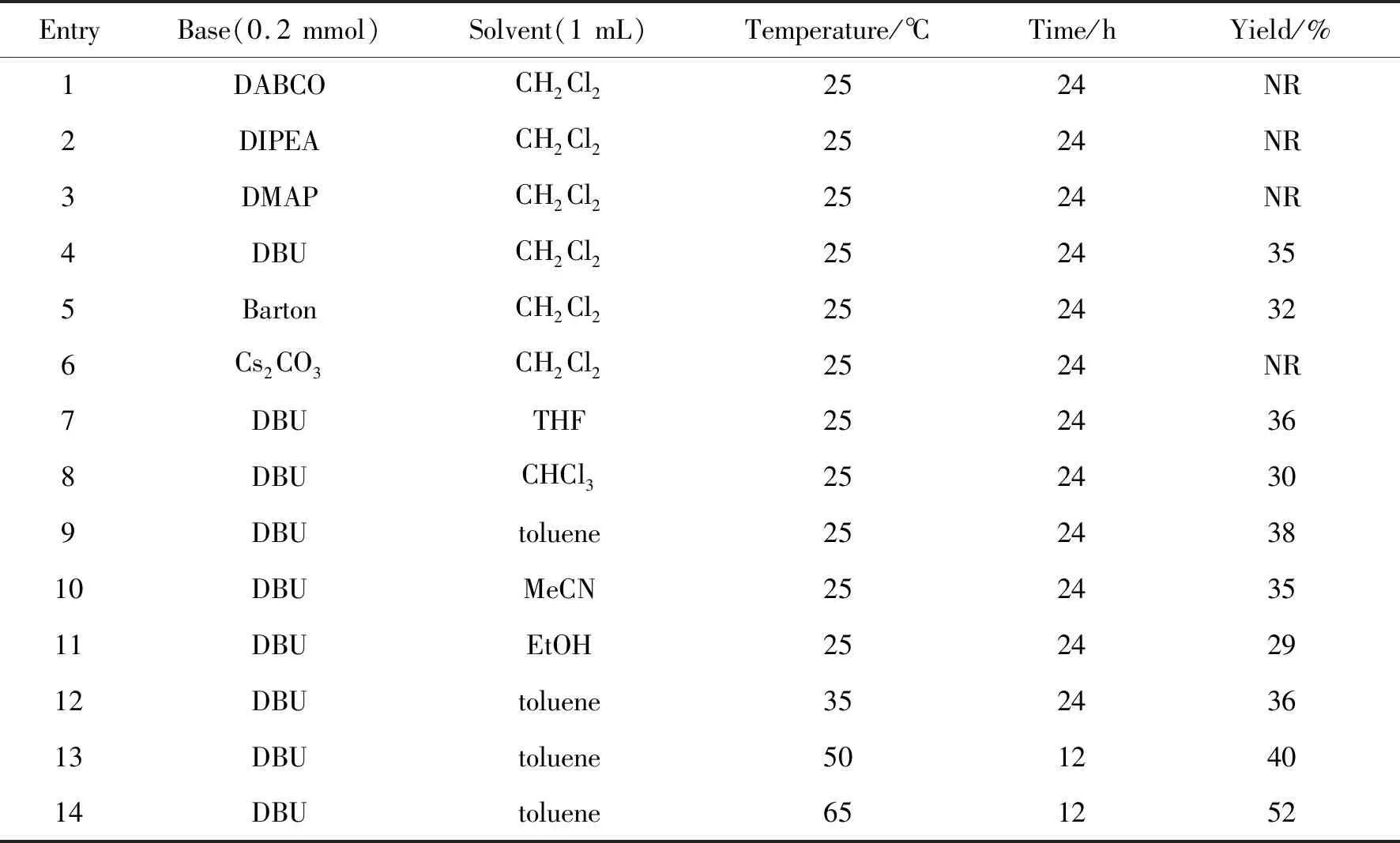
表1 一锅法合成硫代黄酮衍生物反应条件的优化Table 1 Optimization of reaction conditions for one-pot synthesis of thioflavone derivatives
由表1可知,在所筛选的碱中除了DBU和巴顿碱外,其余无机碱Cs2CO3和有机碱DIPEA、DMAP等均不能催化反应使其得到目标产物,但反应无法获得较高收率(Table 1,Entry 1~6)。由此开展了反应溶剂的优化,选择的溶剂包含乙腈、四氢呋喃、芳烃、乙醇。由结果可知反应溶剂对反应的效率影响较小,收率仍旧较低,选择了收率相对最高的甲苯作反应溶剂继续调整其他条件(Table 1,Entry 7~11)。当逐渐提高反应温度后,可明显缩短反应时间,催化活性提高反应收率上升(Table 1,Entry 12~14)。综上,以甲苯作溶剂,2-硝基苯并呋喃1a与邻巯基苯甲醛2在DABCO(0.1 eq.)和DBU(2 eq.)催化下,65 ℃反应12 h,能以52%的收率得到产物3a。
2.2 底物的扩展
在最优反应条件下,对反应底物进行扩展,实验结果如Scheme 1所示。由结果表明,2-硝基苯并呋喃苯环上不同位置被吸电子基(如-F,-Cl,-Br,-NO2)时,除硝基取代需延长反应时间外,其余取代的底物反应12 h即可以43%~67%产率获得目标产物(3b~3e)。R为供电子基(如-Me,-OMe,t-Bu)时,以42~47%的收率得到目标产物(3f~3h)。对于双取代底物,如5,7-二溴和5-溴-7-甲氧基底物,均能参与反应,分别以52%的收率和55%的收率得到产物3i和3j。当底物为大位阻的萘并呋喃时,由于位阻原因只能以45%收率获得相应产物3k。可见各种类型的基团在此反应中能够很好的兼容,这为产物的后续衍生提供了便利。
开发了以2-硝基苯并呋喃与邻巯基苯甲醛为底物,通过串联去芳香化[4+2]环加成/消除/氧化一锅法反应得到硫代黄酮衍生物的合成路线。反应具有较好的普适性,不同电性取代的2-硝基苯并呋喃均能顺利的获得相应产物。该方法为合成一种新的硫代黄酮衍生物提供了一种简便可行的途径。
