IL2RG基因新突变致X-连锁联合免疫缺陷1例*
2021-03-13陈琼华郑敬阳林春燕曾丽娥林印涛福建省泉州市儿童医院362000
陈琼华 郑敬阳 张 谭 林春燕 曾丽娥 林印涛 福建省泉州市儿童医院 362000
X连锁严重联合免疫缺陷(X-linked severe combined Immunodeficiency, X-SCID)是由编码普通γ链(IL2RG)的基因突变引起的一种罕见的危及生命的疾病。目前国内有散发的病例报道,本文通过1例IL2RG基因新突变致 X-连锁联合免疫缺陷的报道,希望增加对非典型X-SCID的认识,有助于早期诊断该类疾病。
1 病历资料
首次住院情况:患儿男,6个月,因“咳嗽、气喘10余天,发热1d”于2018年3月收入我院呼吸科住院。入院前10余天,无明显诱因出现咳嗽,呈阵发性,咳时有痰,伴气喘,病初第2天于外院住院,考虑“肺炎”,曾予“头孢哌酮舒巴坦”抗感染,丙种球蛋白免疫支持,雾化等治疗8d,期间咳嗽有所好转,痰声仍明显,时有气喘。入院前1d,患儿始出现发热,热峰39.0℃,退热后体温可降至正常,但仍反复,共3次,转诊我院,拟“肺炎”收住院。患儿系G2 P2,37+3周因“羊水少”剖宫娩出,否认窒息抢救史,出生体重2.35kg,有一姐姐,7岁,体健。患儿平素体质一般,入院前20余天,曾诊断“重症肺炎”,于外院PICU治疗14d好转出院。有湿疹史,生后至今喘息3次,首次发作为1个月前,当时无伴发热,第2次,第3次为发热伴喘息,此次入院为第3次发作。
入院查体:T 37.0℃,P 154次/min,R 50次/min,SpO299%,Wt 7.5kg,神志清楚,眉毛可见黄色皮屑,可见卡疤,咽充血,呼吸稍促,两肺闻及固定中细湿啰音及哮鸣音,心律齐,未闻及杂音,腹软,肝脾肋下未触及,肢端温。
辅助检查:入院第1天血常规:WBC 4.3×109/L、NE 36.3%、LY 52.2%,NEUT# 1.57×109/L,LYM# 1.57×109/L,HGB 114g/L、PLT 244×109/L,CRP 1.07mg/L,体液免疫:IgG 12.6g/L,IgA 0.26g/L,IgM 0.76g/L,C3 0.98g/L,C4 0.25g/L。胸部CT:左肺炎,右侧气管性支气管,右侧中间支气管狭窄。支气管镜:(1)支气管内膜炎(右上叶、右中叶、右下叶、左下叶基底段)。(2)右侧气管性气管。(3)右中间支气管,右中叶支气管狭窄(见图1)。痰培养(肺泡灌洗液):肺炎链球菌,青霉素G敏感,阿莫西林耐药,头孢噻肟耐药,红霉素耐药。入院第21天血常规:WBC 2.9×109/L,NEUT 50.6%,LYM 40.8%,NEUT# 1.48×109/L,LYM# 1.19×109/L,HGB 131g/L,PLT 240×109/L。入院第25天血常规:WBC 6.0×109/L,NE 16.4%,LY 74.5%,NEUT# 0.98×109/L,E 0.7%,LYM# 4.44×109/L。
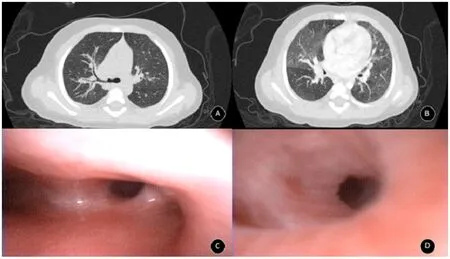
图1 IL2RG基因突变致 X-连锁联合免疫缺陷患儿的胸部CT及支气管镜下表现A.右侧中间支气管狭窄 B.小气道通气不均 C.右侧支气管性支气管 D.右中叶开口稍狭窄,炎性改变
入院第1天予“头孢哌酮舒巴坦”抗感染,“甲泼尼龙”抗炎,雾化止咳;入院第3天行支气管镜肺泡灌洗,可见:右侧气管性支气管,右中间支气管,右中叶支气管狭窄;第5天甲泼尼龙减量;第8天根据药敏加用青霉素抗感染;第9天停用甲泼尼龙;第12天停用青霉素,改用万古霉素加强抗感染;第18天,出现咳嗽喘息反复,伴发热,热峰38.4℃,腹泻,每日7~8次黄色稀水便,予改用氨基酸奶粉,改用头孢曲松联合万古霉素抗感染;第20天停用抗生素,加强雾化;第22天体温正常,咳喘减轻,精神好转,腹泻改善;第24天,活动后气喘,肺部闻及少许哮鸣音,好转出院。鉴于患儿病情迁延,生后反复喘息,有1次重症肺炎,间断发热,有湿疹史,父亲有喘息史。需注意排除免疫缺陷、纤毛运动障碍、囊性纤维化、哮喘等疾病,获得父母知情同意后,对患儿及父母行全外显子检测。基因结果回报:IL2RG基因的一个变异c.759(exon6)G>T(见图2),为半合子,符合X染色体隐性遗传疾病发病机制;符合先证者及其家系成员表型及基因型的共分离。出院后予氟替卡松,硫酸沙丁胺醇气雾剂吸入,患儿喘息有缓解。1个月后复查血常规:WBC 5.6×109/L,NE 74.8%,LY 19.1%,HGB 118g/L,PLT 206×109/L,NEUT# 4.22×109/L,LYM# 1.08×109/L。3个月后停用吸入激素。
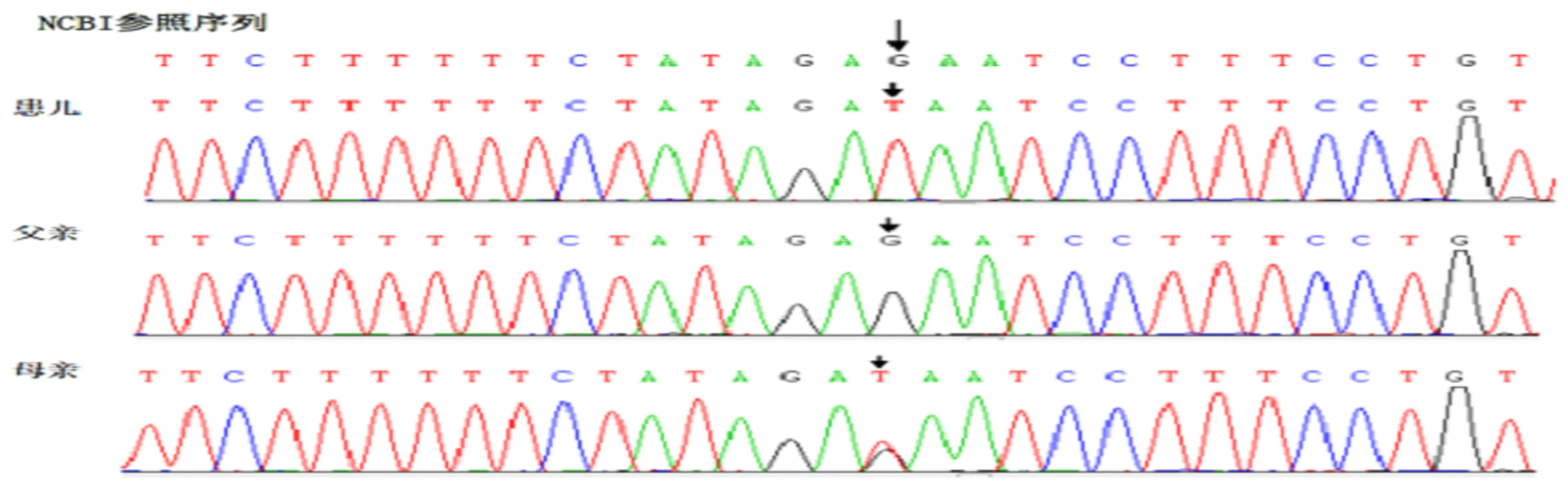
图2 IL2RG基因突变致 X-连锁联合免疫缺陷患儿及父母基因检测
第2次住院:首次出院3个月后再因“发热2d”住我院,期间伴有腹泻症状,查血常规:WBC 3.4×109/L,NE 26.9%,NEUT 0.91×109/L, LY 62.5%,LYM# 2.21×109/L,HGB 115g/L,PLT 130×109/L。予退热,补液好转出院。
第3次住院:首次出院9个月后再因“发热咳嗽”住院,查血常规WBC 7.3×109/L,NE 41.4%,NEUT# 3.02×109/L,LY 45.8%,LYM# 3.34×109/L,HGB 120g/L,PLT 173×109/L。抗感染,雾化好转出院。
第4次住院:首次出院10个月后,患儿已1岁5月龄,再次因:咳嗽7d,发热2d,加重伴气喘1d住院。入院前1d血常规:WBC 23.9×109/L,NE 15.8%,LY 77.2%,E 1.6%,NEUT 3.77×109/L,LYM 18.41×109/L,体液免疫:IgG 4.17g/L,IgA 0.63g/L,IgM 1.17g/L,C3 0.98g/L,C4 0.30g/L,入院第6天血常规:WBC 29.9×109/L,NE 15.3%,LY 81.2%,HGB 131g/L,PLT 493×109/L,LYM 24.3×109/L。外周血涂片:异型淋巴细胞3%,成熟红细胞形态未见明显异常。细胞免疫:总T细胞(CD3)百分率62%,Ts/Tc细胞(CD3+CD8+)百分率24%,Th/Ti细胞(CD3+CD4+)百分率34%,CD4+/CD8+比值:1.42,NK细胞(CD3-CD16+CD56+)百分率9%,总B细胞(CD19+)百分率25%。入院第10天血常规11.4×109/L,NE 27.3%,NEUT# 3.31×109/L,LY 69.7%,LYM# 9.95×109/L,HGB 135g/L,PLT 269×109/L。痰呼吸道病原体(荧光PCR法):百日咳杆菌(CT值19.82)、腺病毒(CT值16.77)、肺炎链球菌(CT值24.93)、呼吸道合胞病毒(CT值16.27)阳性。
入院先后予阿莫西林舒巴坦、头孢哌酮舒巴坦、万古霉素、红霉素抗感染、甲泼尼龙抗炎,以及雾化,期间曾出现体温反复,住院19d好转出院。出院后予氟替卡松、硫酸沙丁胺醇气雾剂吸入,序贯口服阿莫西林克拉维酸钾及阿奇霉素,口服甲泼尼龙逐步减量应用1周后予停用,继续门诊随访。
2 讨论
IL2RG定位于X染色体q13.1,有8个外显子,4 145个碱基对,编码全长为369个氨基酸的蛋白质。IL-2R信号在T细胞成熟、活化和体内平衡中必不可少,IL-2通过与免疫细胞膜上的IL-2R结合而发挥作用,IL-2Rγ链不仅是维持IL-2R复合体完整性及IL-2/IL-2R复合物内化所需,而且是联系胞膜表面细胞因子结合区域与下游胞内内信号传导分子的纽带。由于细胞免疫和体液免疫缺陷,受感染婴儿生后3~6个月往往会出现严重机会性感染。T细胞和自然杀伤细胞显著减少;B细胞数量正常或增多,但功能异常,导致免疫球蛋白产生减少和类别转换障碍。
1993年,Noguchi等[1]报道编码IL-2Rγ的基因IL2RG突变是X-SCID的主要病因。本病的发病率为1/75 000~100 000名新生婴儿,IL2RG突变占SCID患者40%~50%。世界上已经报道 264种IL2RG突变引起的SCID,突变热点集中在cDNA 690-691以及cDNA 879的CG二核苷酸处。目前尚无确切的发病率统计,已经证实20余种基因突变导致不同临床表型和免疫表型的SCID,分别是IL2RG(IL-受体γ链)、ARTEMIS、RAGI、RAG2、ADA、CD45、JAK3和IL7R等[2-3]。本例患儿IL2RG突变位于第6个外显子,第759位子碱基G错义突变为碱基T,导致第253为谷氨酸突变为天冬氨酸,患儿父亲为正常野生型,母亲为携带者,此种突变类型目前尚未见报道。
X-SCID患儿发病较早,3~6月龄出现严重反复感染,典型表现还包括体重不增、长期念珠菌感染和慢性腹泻。常出现条件致病菌,如:卡氏肺孢子虫、曲霉菌、李斯特菌和军团菌,也易并发病毒感染如:巨细胞病毒、水痘病毒、疱疹病毒、麻疹病毒和腺病毒,常可引起进行性、致命性感染。婴儿期有严重的皮肤感染,皮疹,类似于湿疹,但分布更广泛。刘钋宁等[4]曾总结4例基因确诊病例,均表现为严重肺部感染且胸部X线片均提示无胸腺或胸腺显著缩小,3例接种卡介苗后出现结核分枝杆菌感染,2例有家族病史,4例淋巴细胞绝对计数均减少。不同基因变异导致不同形式的免疫缺陷和非典型临床表现。例如,RAG或IL2RG基因的低形态突变可导致OMENN综合征,这是一种具有残余T细胞的免疫缺陷疾病,其严重程度低于典型的SCID。Mou等[5]曾有报道1例IL2RG基因第6号外显子第52位核苷酸缺失突变所致X-SCID的非典型表现,为1名15月龄男孩,反复发热,肺炎,持续性肠道感染,因反复感染和腹泻而死于营养不良,这是免疫功能不全累积的次要后果,与典型SCID中突发死亡和严重感染的病例有所不同。流式细胞仪分析显示,该患者T细胞明显减少,但仍有完整的增殖和广泛的TCR谱,在淋巴细胞中意外发现不成熟的NK细胞比例增加。
本例临床表现似乎不典型,出生后不久也有湿疹表现,程度不剧,在6个月就诊查体仍可见眉尖黄色皮屑,生后5月龄始有反复喘息,曾伴有腹泻症状,但经饮食调整后有所改善,右侧气管支气管畸形,先后多次因反复感染于我院住院,先后检出肺炎链球菌、百日咳杆菌、腺病毒、呼吸道合胞病毒。中性粒细胞绝对值计数在感染时有一过性缺乏,但经治疗后又恢复正常,淋巴细胞绝对值始终在正常范围,细胞免疫中T细胞,B细胞,NK细胞比例正常范围,体液免疫血清免疫球蛋白浓度正常,但可能与6月龄时接受免疫球蛋白治疗有关。然而,基因分析本身不足以进行诊断,必须与细胞功能分析相结合,才能将遗传缺陷与免疫表型变化和变异临床表现联系起来。还有,本例患者还太小,无法评估该突变对机体全部的影响,但已知随着年龄增长,该病有可能出现进行性加重的免疫缺陷临床表现,还需更长时间追踪随访。
SCID的根本治疗是免疫重建,造血干细胞移植和基因治疗。治疗重点在于结合病原学合理运用抗生素,隔离患儿于相对无病原环境,对SCID或疑似SCID患儿运用丙种球蛋白替代治疗,直至接受骨髓移植。来自同一同胞捐赠者的造血干细胞移植有效,但只有不到20%的患者可以获得,而来自其他捐赠者的移植会增加与移植物抗宿主病和不完全免疫重建的风险。基因治疗是移植自体干细胞/祖细胞,这些自体干细胞/祖细胞经过逆转录病毒转导了治疗基因,已经成功的对一些患者进行免疫重建,但这种治疗方法仅应用于不能进行骨髓干细胞移植或骨髓干细胞移植失败的患者,虽然基因治疗潜力巨大,但颇具风险。目前最新报道[6]慢病毒载体基因治疗结合低暴露、靶向二甲磺酸丁酯治疗对新近诊断为SCID-X1婴儿的低毒性,导致转导细胞的多系植入,功能性T细胞和B细胞重建以及16个月中位随访期间NK细胞计数的正常化,有望实现突破。
综上所述,本例患儿主要表现反复喘息,右侧气管性支气管,多种病原体感染,湿疹样皮肤损害,腹泻,一过性中性粒细胞减低,有可能是一种新的X连锁免疫缺陷病的“减弱表型”,多数该类患儿治疗困难,及时识别及治疗可以增加患儿的存活率,基因检测有助于早期诊断这类疾病。
