儿童急性髓系白血病合并糖原累积症1例病例报告并文献复习
2021-03-12张然然蔡玉丽竺晓凡杨文钰
孙 雨 张然然 蔡玉丽 竺晓凡 杨文钰
1 病例资料
男,5岁。因“间断发热10余 d,发现血象异常6 d”于2020年6月在中国医学科学院血液病医院(我院)就诊。患儿于2020-6-18无明显诱因出现发热,伴全身多发皮肤紫癜,外院查血常规示:WBC 96.0×109·L-1,Hb 85 g·L-1,PLT 33×109·L-1,幼稚细胞占65%。患儿既往体健,否认外院住院史。个人史:足月剖宫产第1胎,出生体重3.3 kg,生后有轻度新生儿黄疸,自行消退,生后母乳喂养,6月龄添加辅食,1岁半断奶,生长发育同正常同龄儿。家族史:父母体健,非近亲结婚,无家族及遗传病史。
体格检查:中度贫血貌,右下肢可见散在瘀斑,颈部可扪及3个肿大淋巴结,约黄豆大小,边界清楚,活动度可。心肺未见异常,肝脾肋缘下未触及,四肢肌力正常。
实验室检查:血生化 LDH 993(参考值0~247)U·L-1,CK 394(参考值0~171)U·L-1,CKMB 23.6(参考值<24)U·L-1,无机磷1.8(参考值1.3~2.3)mmol·L-1,钙2.7(参考值2.2~2.7)mmol·L-1。骨髓MICM分型:骨髓细胞形态示,增生活跃,G=0.5%,E=0;可见约81.5%的原始细胞,考虑为髓系来源(图1A)。染色体:46,XY,add(1)(p34),del(5)(q31q35),add(15)(q22),inc[20](图1B)。骨髓组织细胞化学染色3项:特异性酯酶(CE)阳性率15%,阳性指数21(图1C),过氧化酶染色(MPO)阳性率96%,阳性指数108,幼稚细胞PAS染色阳性率94%,阳性指数94(图1D)。提示:原幼单核细胞比例增高。白血病免疫分型:异常细胞群占有核细胞的64.7%,表达MPO、CD13、CD33、CD38、CD123、CD56,部分表达CD34,弱表达CD11b、CD64,不表达TDT及其他髓系、淋系标志,符合儿童急性髓系白血病(AML)表型。血液系统疾病基因突变筛查:FLT3/ITD,等位基因比例0.66%。
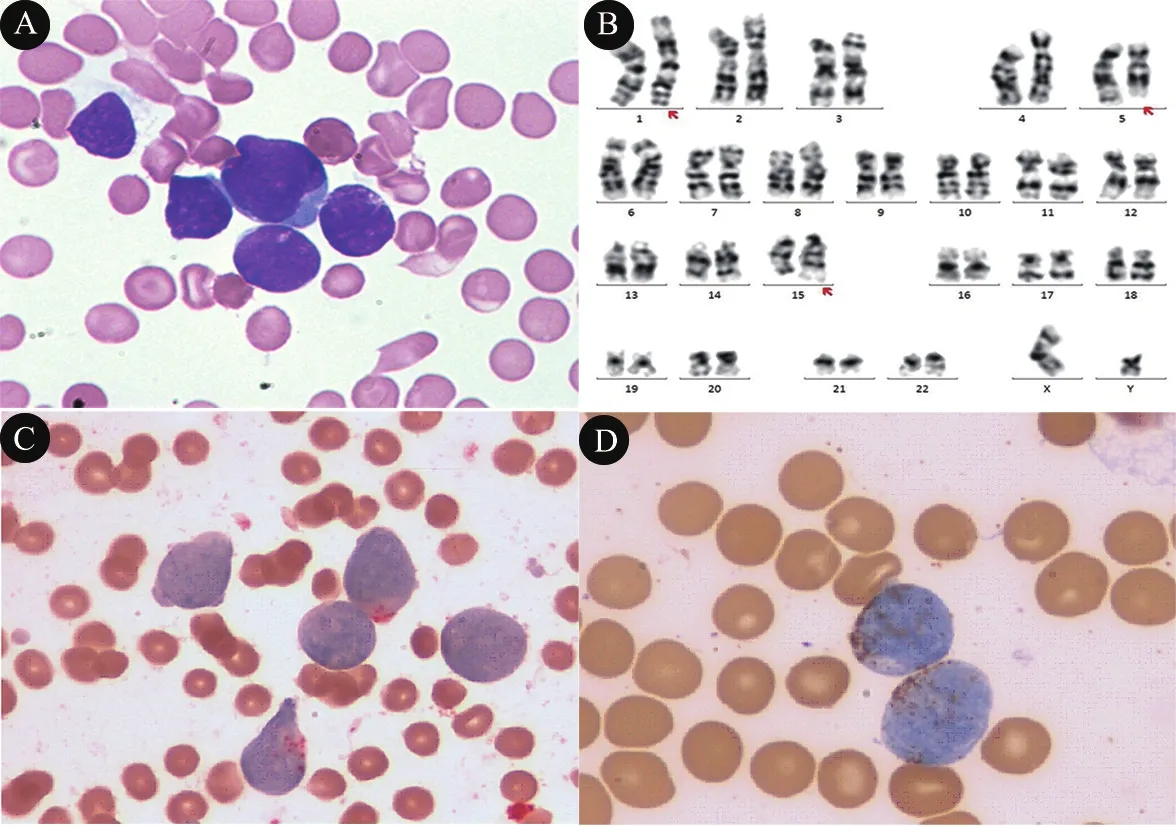
图1 患儿骨髓各项资料
根据《血液病诊断与疗效标准》[1]明确诊断AML-M5(FLT3-ITD突变,高危组)。予IAE(伊达比星、依托泊苷、阿糖胞苷)诱导方案化疗,依托泊苷(VP-16)150 mg·m-2,d 1~5;伊达比星(IDA)8 mg·m-2,d 6~8;阿糖胞苷(Ara-c)200 mg·m-2,d 5~11。血常规恢复后复查骨髓细胞形态及残留均阴性(-)。FLT3-ITD等位基因比例0。后予巩固治疗化疗方案:①Ara-c 3 g·m-2,q12h,d 1~3;②IDA 10 mg·m-2,d 1,Ara-c 3 g·m-2,q12h,d 1~3)。脑脊液常规、生化、流式均未见异常,化疗后骨髓各项指标变化见表1。
因患儿初诊有CK升高,在初始诱导化疗期间持续上升,最高3 942 U·L-1,CKMB 30.3 U·L-1,患儿无肌痛、肌无力相关症状,肌红蛋白(MYO)、肌钙蛋白T(TNT)、尿常规、肾功能及心电图、心脏彩超检查未见异常,予以静滴左卡尼汀并口服辅酶Q10治疗后CK逐渐下降至263.1 U·L-1,巩固治疗化疗方案①结束后CK再次升高达7 041 U·L-1,CKMB 68.4 U·L-1,MYO 225 ng·ml-1,TNI在正常范围,肌电图、心脏彩超未见异常,予口服卡托普利、辅酶Q10后CK下降至603.4 U·L-1,CKMB降至20.1 U·L-1。巩固治疗化疗方案②前患儿CK再次升至3 360.6 U·L-1,CKMB升至40.6 U·L-1,化疗期间给予静滴左卡尼汀、口服辅酶Q10后CK逐渐降至166 U·L-1,CKMB降至24 U·L-1(图2)。因患儿反复不明原因CK、CKMB及MYO升高,均在化疗期间或结束后,不能排除与化疗相关,查阅相关资料后,为排除先天代谢性疾病行肌病相关全外显子测序,患儿及父母基因高通量测序示,患儿PYGM基因有1个纯合突变c.1142T>C(p.L381P)。经家系验证分析,父母均为PYGM基因杂合突变(c.1142T>C)。患儿符合“糖原累积症(GSD)V型”的诊断标准[8]。因家长考虑后续治疗费用较高,未再入院治疗。
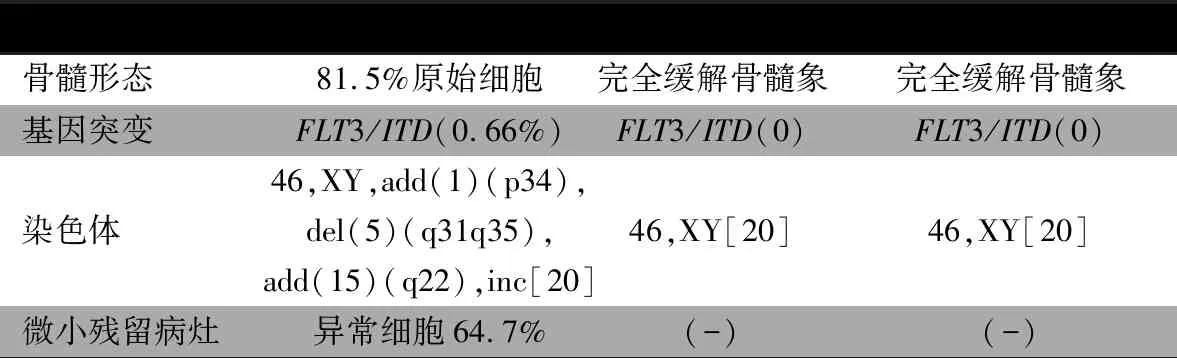
表1 患儿化疗后骨髓各项指标变化情况
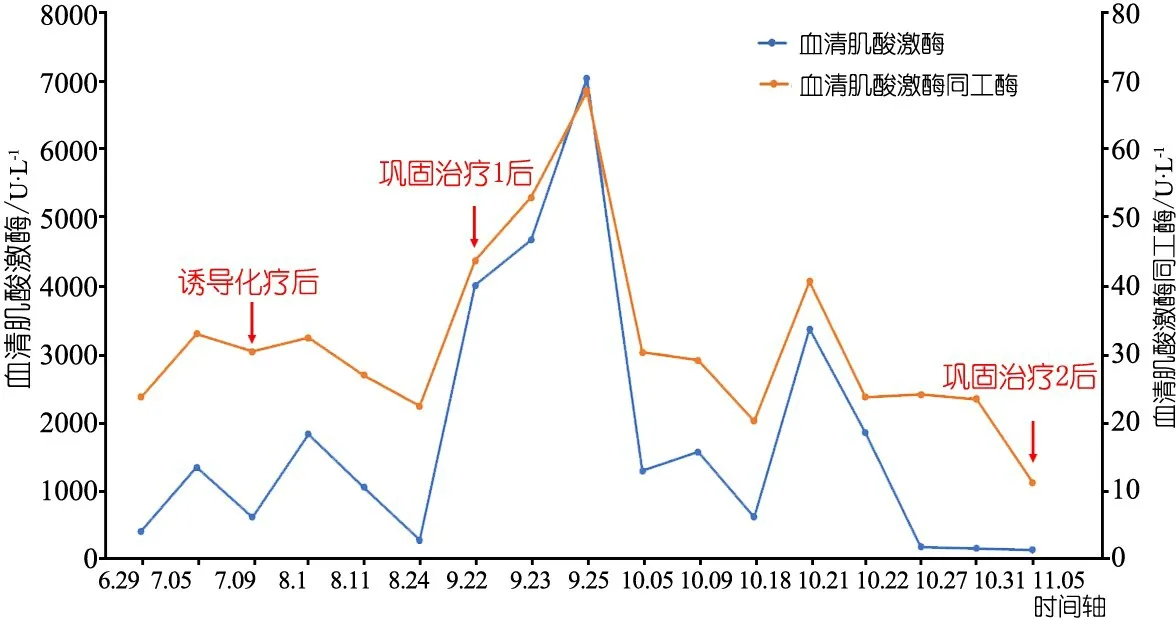
图2 患者血清肌酸激酶和结算集美同工酶变化趋势
2 讨论
AML约占儿童急性白血病的15%,临床预后受多种因素影响,如年龄、细胞遗传学,分子生物学等。本文病例存在FLT3-ITD基因突变。FLT3-ITD是Ⅲ型受体酪氨酸激酶家族成员,位于人体13号染色体长臂,可编码FLT3蛋白,此类突变约占儿童AML的7.8%,低于成人AML,具有初诊外周血WBC计数高、临床预后差、诱导缓解率低、易复发等临床特征。Port等[2]研究显示有FLT3-ITD突变的AML患者的总体生存率(OS)和无复发生存率(RFS)显著降低。目前已有针对FLT3受体激酶的抑制剂,有研究[3]表明在诱导和巩固治疗中加入Midostaurin可显著增加FLT3-ITD突变的AML患者的RFS。与常规化疗相比,Stone等[4]在研究中发现在患者完全缓解后进行造血干细胞移植(HSCT),能使患者的长期生存率达到70%,同样有研究[5]数据表明存在FLT3-ITD的AML患者如不进行HSCT,其OS及RFS相较于进行HSCT者更差。此类突变预后较差,复发风险较高,即使完全缓解,复发风险率也高达70%,故化疗缓解后应尽早进行allo-HSCT。
糖原累积病(GSD)根据酶缺陷或转运体的不同可分为13种亚型,其中以肝脏和肌肉最易受累。GSD V型,也被称为麦卡德尔病,是一种常染色体隐性遗传的代谢性肌病,由于11号染色体上PYGM基因突变导致肌磷酸化酶活性明显减低或缺失,使糖原支链的α-1,4葡萄糖苷键不能被水解生成1-磷酸葡萄糖,影响糖原分解和ATP的产生,糖原在肌纤维内大量堆积,此病仅累及肌肉。其特点是运动不耐受,表现为快速疲劳、肌痛和肌肉痉挛,经休息后可明显好转。症状通常由等长运动或持续有氧运动引起肌无力患者约占25%,多见于年龄较大者。约50%的患者有反复发作性肌红蛋白尿,最终可能导致急性肾功能衰竭[6]。此外,有一些不常见的临床表现,如咀嚼困难、吞咽困难、口腔运动功能障碍等,多见于年龄较小的患者。其他罕见表现有自发性筋膜室综合征[7]或后颈部肌肉挛缩[8]。实验室检查90%患者可见CK呈轻至中度升高,可有一定波动性。该病发病年龄10岁左右,但通常在成年后才得以诊断。本文病例仅有CK增高,有文献报道[9],在婴儿期和青春期前可仅表现为无症状的CK增高,最高可达17 000 U·L-1。GSD V型诊断的首选方法是基因检测,目前尚无有效的治疗方法。
据文献报道[10],AML合并GSD多为GSD 1b型。GSD 1b是由于染色体11q23上的葡萄糖-6-磷酸转移酶基因突变引起,该病表现为发育落后、肝肿大、低血糖、乳酸中毒以及中性粒细胞减少症导致的感染。有研究表明,GSD 1b型导致的中性粒细胞减少症长期使用粒细胞刺激因子(G-CSF)后可诱发AML/MDS,全世界目前共报道5例[11-15](表2),其中3例为儿童,2例为成人,1例在使用G-CSF之前诊断AML,4例为长期使用G-CSF之后诊断,其使用G-CSF的中位年龄为12.5年,2例死亡,3例经HSCT后生存良好。有研究证实,GSD 1b型患者长期使用G-CSF可能导致骨髓造血细胞过度增生和端粒缩短,可能导致白血病的发生[16]。

表2 目前已报道的GSD合并AML临床特征及转归
有学者提出,因GSD 1b患者接受高剂量的G-CSF治疗可能会增加白血病的风险,故应减低用药剂量[17],文献复习的5例患者在长期接受G-CSF治疗后并发AML。有研究者提出在合并中性粒细胞减少的患者中可能有一种突变的G-CSF受体,发出异常增殖信号,导致幼稚细胞增殖,这一缺陷与7号染色体异常有关[18-19]。G-CSF在白血病发生中的作用机制尚不清楚,建议GSD 1b患者监测血液恶性肿瘤的发展情况。对于伴有中性粒细胞或全血细胞减少的GSD 1b患者,无论是否接受G-CSF治疗,都应尽早行骨髓检查,以免延误诊断及治疗。一经诊断后应尽早行HSCT。
目前国内外文献未见AML合并GSD V型的病例报道,发病机制尚未清楚,导致GSD V型的PYGM基因突变并无导致AML的风险。PYGM基因编码肌磷酸基酶,迄今已报道147个致病突变和39个多态性,外显子1和17是PYGM的热点突变,其中50%为无义突变,而c.148C>T(通常称为p.R50X)在大多数研究群体中是最常见的突变[20]。本文患儿早期着重对AML的诊治而忽略了GSD V型的识别诊断,初诊时即有CK轻度增高,心电图、心脏彩超未见异常,而随着后期予对症治疗,CK呈现降低趋势,之后又无明显诱因增高,且与化疗无明显相关性,由此考虑患儿有其他原发疾病,通过基因检测最终诊断GSD V型,根据以往研究发现此病可导致CK增高,且可呈一定波动。
根据本文患儿引发对于GSD V型疾病的关注,如果患儿出现CK无明显诱因升高,应及时询问病史,查体并完善相关检查,查阅相关资料是否存在先天代谢相关疾病,警惕合并其他疾病的可能,GSD V型的最终诊断依据肌肉活检或基因突变检测。由于GSD V型较为罕见,且无有效治愈措施,高碳水低脂化合物饮食及有氧运动对症状及肌肉功能的改善可能有帮助。
