熊果酸磷脂复合物纳米结构脂质载体的制备及其体内药动学研究
2021-03-09张艳慧李淑荣
张艳慧,丁 玉,李淑荣
(1.黄河科技学院,河南郑州 450005;2.河南中医药大学第一附属医院,河南郑州 450000)
熊果酸属于五环三萜类化合物,主要存在于枇杷叶、熊果果实、女贞叶、毛泡桐叶等植物部位中[1],具有镇静、抗炎、抗氧化、调血脂、抗肿瘤、保肝、抗粥样动脉硬化等多种药理作用[2-5],对人卵巢癌耐顺铂SKOV3/DDP 细胞裸鼠移植瘤具有较好的抑制活性[6],其机制可能与抑制抗凋亡因子Bcl-2 表达有关,故备受科研工作者的关注[7-8]。但熊果酸溶解性较差[9],口服生物利用度较低,限制了其药效的充分发挥及在医学领域中的应用。
磷脂复合物是药物和磷脂在一定条件下通过氢键、范德华力等形式结合而成一种复合物[10-11],可改善药物溶解性能,并促进其吸收,提高其生物利用度,但它易受胃肠道酶解、pH 等因素的影响,使得生物利用度提高程度有限[12]。纳米结构脂质载体是在固体脂质纳米粒基础上发展起来的一种新剂型,具有稳定性好、载药量高等优势,将磷脂复合物包裹于其中时可对前者提供保护作用,从而增加其稳定性。因此,本实验制备熊果酸磷脂复合物纳米结构脂质载体,并考察其体内药动学,以期为相关研究提供参考。
1 材料
1.1 仪器 Agilent 1260/6310 型高效液相色谱-质谱联用仪(美国Agilent 公司);AB265-S/FACT 型电子天平[梅特勒托利多仪器(上海)有限公司];85-2 型数显恒温磁力搅拌器(常州市亿能实验仪器厂);SHZ-82 型空气恒温台式振荡器(常州金坛良友仪器有限公司);JC-S12/S24 型水浴氮吹仪(青岛聚创世纪环保科技有限公司);XW-80A 型漩涡混合器(上海医科大学仪器厂)。
1.2 试剂与药物 熊果酸原料药(批号PT20171019,西安飞达生物科技有限公司);熊果酸对照品(批号YY20160525,纯度99.2%,上海源叶生物科技有限公司);甘草次酸对照品(批号110123-201608,中国食品药品检定研究院);大豆卵磷脂(批号PC-98T,上海辅必成医药科技公司);单硬脂酸甘油酯(Kolliwax GMS,德国巴斯夫公司)。四氢呋喃为色谱纯(德国Merck 公司)。
1.3 动物 SD 大鼠购自上海斯莱克实验动物有限公司,雌雄兼具,体质量(300±20)g,动物生产许可证号SCXK(沪)2016-0002。
2 方法与结果
2.1 熊果酸含量测定
2.1.1 色谱条件 Agilent Extend C18色谱柱(250 mm×4.6 mm,5 μm);流动相乙腈-0.1 mol/L磷酸(85 ∶ 15);体积流量1.0 mL/min;柱温35 ℃;检测波长210 nm;进样量20 μL。
2.1.2 方法学考察 精密称取熊果酸对照品20.0 mg,溶于50.0 mL 乙腈中,即得400.0 μg/mL母液,精密量取适量,流动相依次稀释至200.0、100.0、50.0、25.0,10.0、2.0 μg/mL,在“2.1.1”项色谱条件下进样测定。以峰面积(Y)对熊果酸质量浓度(X)进行回归,得方程为Y =0.039 6X -0.007 4(r =0.999 8),在2.0~200.0 μg/mL范围内线性关系良好。
取200.0、50.0、2.0 μg/mL 对照品溶液,在“2.1.1”项色谱条件下各进样测定6 次,测得日内精密度RSD 分别为0.09%、0.13%、0.17%;各连续进样测定6 d,每天1 次,测得日间精密度RSD 分别为0.31%、0.24%、0.69%,表明该方法精密度良好。取熊果酸对照品10 mg,溶于100 mL乙腈中,超声溶解,即得供试品溶液,于0、2、4、6、12、24、48 h 在“2.1.1”项色谱条件下进样测定,测得其峰面积RSD 为0.58%,表明溶液在48 h 内稳定性良好。取空白纳米结构脂质载体混悬液,分别制备含10.0、25.0、50.0 μg/mL 熊果酸的样品溶液,在“2.1.1”项色谱条件下进样测定6 次,测得平均加样回收率分别为100.57%、99.12%、99.34%,RSD 均小于 1.12%。取2.0 μg/mL对照品溶液,将“2.1.1”项下流动相逐步稀释,测得定量限(S/N=10)为5.0 ng/mL,检测限(S/N=3)为2.0 ng/mL。
2.2 熊果酸磷脂复合物制备及其复合率、溶解度测定 取熊果酸原料药、大豆卵磷脂各50 mg(摩尔比1 ∶1.1),置于三角烧瓶中,加入35 mL 四氢呋喃,45 ℃下恒温搅拌4.0 h,40 ℃下减压旋蒸除去四氢呋喃,即得。
称取熊果酸、大豆卵磷脂适量(质量均为X0),同法制备熊果酸磷脂复合物,加入适量石油醚振荡溶解,0.22 μm 微孔滤膜过滤,除去游离的熊果酸,收集滤液,40 ℃下减压旋蒸除去石油醚,收集固体,溶于10 mL 乙腈中,在“2.1.1”项色谱条件下进样测定,计算熊果酸质量(X1)和复合率,公式为复合率=X1/X0×100%,平行3 次,测得其数值均接近100%。
参考文献[13]报道的方法,取过量熊果酸及其磷脂复合物,加入蒸馏水磁力搅拌2 d,经0.45 μm 微孔滤膜过滤,在“2.1.1”项色谱条件下进样测定。结果,磷脂复合物将熊果酸在水中的溶解度由5.36 μg/mL 提高至16.49 μg/mL。同法测定熊果酸在油酸中的溶解度,发现它由0.62 mg/mL 提高至11.52 mg/mL。
2.3 熊果酸磷脂复合物纳米结构脂质载体制备取熊果酸磷脂复合物50 mg、单硬脂酸甘油酯300 mg、油酸60 mg,加入10 mL 乙醇溶解,70 ℃水浴加热,作为油相;称取0.2 g 泊洛沙姆188 溶于20 mL 蒸馏水中,加热至70 ℃,作为水相,设置搅拌速度为800 r/min,将水相逐滴滴入油相中,再将初乳置于冰水混合物中继续搅拌2 h,超声(600 W)处理6 min(每工作2 s 停止3 s),过滤后滴加蒸馏水至20 mL,即得,密封低温保存。以32 mg 磷脂代替熊果酸磷脂复合物(50 mg 磷脂复合物中约含32 mg 磷脂),同法制备空白纳米结构脂质载体混悬液。
2.4 包封率、载药量测定 取1.0 mL 熊果酸纳米结构脂质载体混悬液至超速离心管中,平行2 份,置于高速离心机转子中,10 000 r/min 高速离心45 min,HPLC 法测定滤液中游离药量(m游离)。取1.0 mL,加入适量甲醇后超声处理3 min,定容至10 mL,HPLC 法测定总药量(m总量),计算包封率、载药量,公式分别为包封率= [(m总量-m游离)/m总量]× 100%、载药量= [( m总量-m游离)/(m总脂质+m总量)]×100%,其中m总脂质表示纳米结构脂质载体总质量,平行3 次,测得两者平均值分别为80.54%、3.57%。
2.5 粒径、Zeta 电位测定 取熊果酸纳米结构脂质载体混悬液0.2 mL,加入5 mL 蒸馏水稀释,取适量置于比色皿中,测得其平均粒径为(209.32±4.47)nm,PDI 为0.107 ± 0.011,Zeta 电位为-(10.82±0.42)mV,见图1~2。
2.6 形态观察 将熊果酸磷脂复合物纳米结构脂质载体混悬液适当稀释后,滴加于有支持膜的铜网上,铺展均匀后放置8 min,于5%磷钨酸溶液中浸泡6 min,晾干后在透射电镜(TEM)下观察其形态,结果见图3。由此可知,该制剂呈类球形或球形,纳米粒之间未粘连。

图1 熊果酸磷脂复合物纳米结构脂质载体粒径分布Fig.1 Particle size distribution of nanostructured lipid carriers of ursolic acid phospholipids complex
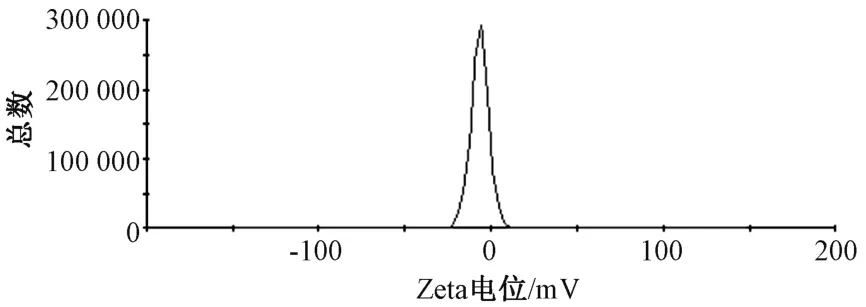
图2 熊果酸磷脂复合物纳米结构脂质载体Zeta 电位Fig.2 Zeta potential of nanostructured lipid carriers of ursolic acid phospholipids complex

图3 熊果酸磷脂复合物纳米结构脂质载体TEM 图Fig.3 TEM image for nanostructured lipid carriers of ursolic acid phospholipids complex
2.7 冻干粉制备及表征 取2 mL 熊果酸磷脂复合物纳米结构脂质载体混悬液至西林瓶中,加入5%甘露醇作为冻干保护剂,振荡一段时间后置于-60 ℃超低温冰箱中预冻2 d,迅速放入冻干机中(初始温度-45 ℃)平衡6 h 后抽真空,保持2 d,4 ℃/h 升至25 ℃,继续保持12 h,即得,其质量分数(以熊果酸计)为1.05%。
2.7.1 粒径、Zeta 电位 取冻干粉约5 mg,加入40 mL 蒸馏水复溶,测得其平均粒径增大至(276.22±7.04)nm,而平均Zeta 电位为-(8.11±0.53)mV,绝对值有所下降。
2.7.2 包封率、载药量 蒸馏水复溶冻干粉后,按“2.4”项下方法测得其平均包封率为73.83%,载药量为3.12%。
2.7.3 形态观察 取冻干粉适量,置于扫描电镜(SEM)下观察,结果见图4,可发现分布于其中的球形纳米粒子。

图4 冻干粉SEM 图Fig.4 SEM image for lyophilized powder
2.7.4 体外释药 1%十二烷基硫酸钠溶液制备熊果酸磷脂复合物纳米结构脂质载体冻干粉混悬液2 mL(含4 mg 熊果酸),置于透析袋(截留分子量8 000~14 000 Da)中;0.5 mL 乙醇溶解熊果酸原料药后,加入1% 十二烷基硫酸钠溶液至2 mL(含4 mg 熊果酸),置于透析袋中。以900 mL 1%十二烷基硫酸钠溶液为介质,在转速100 r/min、温度37 ℃下取样两者各3 mL,同时及时补加3 mL空白介质,测定熊果酸含量,计算其累积释放度,结果见图5。

图5 熊果酸体外释药曲线Fig.5 In vitro drug release curves for ursolic acid
由此可知,熊果酸溶出较快,2 h 内溶出度达95.8%;熊果酸磷脂复合物纳米结构脂质载体冻干粉在前8 h 释药较快,8~36 h 释药较慢。再分别采用Higuchi、Weibull、一级释放模型对其释药过程进行拟合,发现它与Weibull 模型拟合度最高(R2=0.990 6)。
2.8 体内药动学研究
2.8.1 色谱、质谱条件 ZORBAX SB C18色谱柱(2.1 mm×50 mm,1.8 μm);流动相乙腈-0.1%冰醋酸(80 ∶ 20);体积流量0.2 mL/min;柱温30 ℃;进样量5 μL。电喷雾离子源,负离子模式扫描,范围m/z 100~1 000;雾化气压力308.17 kPa;干燥气温度325 ℃;毛细管电压-3 000 V;定量分析离子m/z 469.3(甘草次酸)、 m/z 455.3(熊果酸)。
2.8.2 灌胃液制备 取熊果酸、熊果酸磷脂复合物、熊果酸磷脂复合物纳米结构脂质载体冻干粉适量,加入0.5%CMC-Na 溶液,即得(以熊果酸计,质量浓度为12.5 mg/mL)。
2.8.3 给药、取血、血浆样品处理 12 只大鼠随机分为2 组,实验期间禁食不禁水,按100 mg/kg(以熊果酸计)剂量灌胃给予“2.8.2”项下灌胃液,乙醚麻醉后于0.25、0.75、1、1.5、2、2.5、3、4、6、8、12 h 眼眶静脉丛取血各约0.3 mL,置于肝素化离心管中,用力振荡后3 000 r/min 离心2 min,低温冷冻保存血浆。吸取血浆样品200 μL、内标溶液(精密称取甘草次酸对照品10.0 mg,溶于100 mL 乙腈后得0.1 mg/mL 母液,乙腈稀释至50.0 ng/mL,即得)50 μL、乙酸乙酯1.5 mL,涡旋混匀6 min,5 000 r/min 离心10 min,取出有机相,45 ℃氮气流缓慢吹干后得残渣(控制吹速),加入200 μL 甲醇复溶,涡旋1 min,5 000 r/min 继续离心5 min,转移至内衬管中,在“2.8.1”项条件下进样测定。
2.8.4 线性关系考察 取“2.1.2”项下10.0 μg/mL对照品溶液,乙腈依次稀释至15.0、60.0、120.0、240.0、360.0、480.0、960.0 ng/mL,各取200 μL,加入50 μL 内标溶液,40 ℃氮气流缓慢吹干后加入200 μL 空白血浆,即得血浆对照品溶液,按“2.8.3”项下方法处理,在“2.8.1”项条件下进样测定。以熊果酸质量浓度为横坐标(X),熊果酸、内标峰面积比值为纵坐标(Y)进行回归,得方程为Y =1.307 8X+12.057 4(r =0.990 7),在15.0~960.0 ng/mL 范围内线性关系良好。
2.8.5 专属性试验 取空白血浆、血浆对照品溶液(15 ng/mL)、血浆样品溶液(冻干粉灌胃给药12 h 后),在“2.8.1”项条件下进样测定,结果见图6,可知该方法专属性良好。
2.8.6 提取回收率、基质效应试验 制备30.0、300.0、600.0 ng/mL 质控样品溶液,按“2.8.3”项下方法处理,在“2.8.1”项条件下进样测定峰面积(ρ1);取空白血浆200 μL,除不加内标外按“2.8.3”项下方法处理,于残渣中加入30.0、300.0、600.0 ng/mL 质控样品溶液和内标,在“2.8.1”项条件下进样测定峰面积(ρ2);取30.0、300.0、600.0 ng/mL 质控样品溶液,在“2.8.1”项条件下进样测定峰面积(ρ3),根据公式ρ1/ρ2×100%计算提取回收率,ρ2/ρ3×100%计算基质效应。结果,熊果酸提取回收率分别为88.62%、89.05%、86.66%,内标提取回收率为90.27%;前者基质效应分别为93.59%、92.23%、95.69%,后者基质效应为94.84%。
2.8.7 精密度、稳定性试验 取15.0、240.0、960.0 ng/mL 血浆对照品溶液,1 d 内在“2.8.1”项条件下进样测定6 次,测得其峰面积RSD 分别为12.37%、8.02%、4.97%,表明仪器精密度良好。室温下将血浆样品溶液于0、2、4、6、8、12、24 h 在“2.8.1”项条件下进样测定,测得其峰面积RSD 为7.74%,表明溶液在24 h 内稳定性良好。取15.0 ng/mL 血浆对照品溶液(不加内标),逐步稀释,以S/N≥3 为检测限,S/N≥10为定量限,测得两者分别为1.5、5.0 ng/mL。由此可知,上述结果均符合2020 年版《中国药典》四部9012 项下生物样品定量分析方法验证的指导原则。
2.8.8 结果分析 血药浓度-时间曲线见图7,主要药动学参数见表1。由此可知,将熊果酸制成磷脂复合物后tmax略有延长,但差异无统计学意义(P >0.05),Cmax、AUC0~t、AUC0~∞升高(P <0.01),进一步制成纳米结构脂质载体后tmax更长(P <0.01),Cmax、AUC0~t、AUC0~∞更高(P <0.01);与磷脂复合物比较,纳米结构脂质载体tmax延长(P <0.05),Cmax、AUC0~t、AUC0~∞升高(P<0.01);与原料药比较,磷脂复合物、纳米结构脂质载体相对生物利用度分别增加至2.73、3.95 倍。

图7 熊果酸血药浓度-时间曲线Fig.7 Plasma concentration-time curves for ursolic acid
表1 熊果酸主要药动学参数(,n=6)Tab.1 Main pharmacokinetic parameters for ursolic acid(,n=6)

表1 熊果酸主要药动学参数(,n=6)Tab.1 Main pharmacokinetic parameters for ursolic acid(,n=6)
注:与熊果酸比较,∗∗P<0.01;与熊果酸磷脂复合物比较,#P<0.05,##P<0.01。
3 讨论
难溶性药物进入机体后,需经溶解、溶出、透膜吸收才能进入体循环,从而发挥药效[14-16]。本实验发现,将熊果酸制成磷脂复合物后,该成分在水中的溶解度由5.36 μg/mL 升至16.49 μg/mL,并且Cmax、AUC0~t、AUC0~∞显著升高,相对生物利用度也增加至2.73 倍,可促进药物吸收。
由于磷脂复合物在胃肠道环境中容易发生解离,稳定性也存在一定问题[12,17],故本实验以单硬脂酸甘油酯为固体脂质,油酸为液态脂质,将其进一步制成纳米结构脂质载体,一方面可对包裹于其中的磷脂复合物提供保护作用,有助于提高药物生物利用度;另一方面,可发挥纳米制剂促进药物吸收的作用[12,18]。结果,纳米结构脂质载体的相对生物利用度增加至3.95 倍,Cmax、AUC0~t、AUC0~∞显著升高,tmax显著延长,可能是由于该剂型缓释作用所致,可为今后药效学研究奠定基础[19-21]。
