氮掺杂石墨烯负载金属双原子催化剂对合成气合成C2 氧化物性能调控
2021-03-09常泽海王宝俊凌丽霞章日光
常泽海,王宝俊,凌丽霞,章日光
太原理工大学,省部共建煤基能源清洁高效利用国家重点实验室,山西 太原 030024
合成气(CO 和H2)制C2氧化物被认为是将煤、天然气以及生物质等原料转化为高附加值化学产品的一条切实可行的重要途径[1]。长期以来,合成气制C2氧化物主要采用Cu,Co 和Rh 基传统催化剂[2-4],其中,Cu 对CO 解离能力较弱,容易生成甲醇;Co 有利于CO 活化以及碳链增长,容易生成烃类;Rh 对CO 具有独特的吸附和活化能力,容易生成C2氧化物。这些催化剂普遍存在着原子利用效率低、产物选择性低等问题[5]。
碳基负载金属双原子催化剂[6-9]因其具有原子级分散的金属活性中心,从而表现出独特的催化性能和最大的原子利用率;同时具有较高的金属负载量和易调变的金属活性位点,特别是相邻异核双原子间协同作用能够显著增强催化性能。目前,常用的碳基载体有石墨烯、氮掺杂石墨烯、石墨炔、金属有机骨架衍生的碳质材料、碳纳米管、碳球以及活性炭等[10-13],其中,氮掺杂石墨烯[14-16]因其表面结构易调变、比表面积较大、化学性能优异以及其与负载金属原子适中相互作用等特点而被广泛研究;同时,氮在石墨烯骨架中的掺杂[17-19]不仅增强金属-载体相互作用,而且对金属原子具有电子调控作用,从而提高催化性能。进一步,强的金属-载体相互作用[20]可以避免金属单原子团聚,从而显著提高催化剂稳定性。Xu 等[21]认为氮原子引入碳基载体能够形成更多的缺陷结构,最终改善多孔碳材料的表面特性。Lin 等[22]在活性位点附近引入氮原子作为电子受体,氮原子显著改善活性位点周围的电子分布和局部电场分布,从而提高催化活性。Li 等[23]报道了一种氮掺杂石墨烯负载Co 催化剂,表明单分散的钴原子与氮原子配位,形成稳定和高效的催化活性中心。
从理论上精确地描述活性中心作用下的催化机理是阐明催化剂构效关系的基础。合成气制C2氧化物反应机理复杂,但普遍认为其反应机理主要包括两个关键步骤[24-27]:CO 活化生成CHx中间体和CO/CHO 插入CHx生成C2氧化物。其中CHx中间体生成主要有两条路径:(1)CO 直接活化生成C原子,连续加氢生成CHx;(2)CO 氢助活化生成CHxO 和CHxOH 中间体,其后C—O 键断裂生成CHx。在CHx生成过程中,会伴随着CH3OH 生成,降低CHx选择性。CO/CHO 插入CHx中间体实现C—C 链增长,并生成CHxCO 和CHxCHO 等C2氧化物,该过程会生成甲烷和C2烃类等副产物,降低C2氧化物选择性。因此,合成气高活性高选择性地生成C2氧化物需要CO 高效活化生成CHx和较容易实现C—C 键增长,并抑制甲烷、甲醇以及C2烃类等副产物的生成。
目前,氮掺杂石墨烯负载双原子催化剂上合成气转化反应的研究较少,氮掺杂、同核以及异核活性中心在合成气制C2氧化物反应中的催化作用机制尚不明确。本工作拟构建同核双原子(CuCu@NC,RhRh@NC 和CoCo@NC)和异核双原子催化剂(CuRh@NC,RhCo@NC 和CuCo@NC),采用密度泛函理论计算方法研究不同金属活性中心对CO 活化生成CHx中间体、C2氧化物中C—C 链形成以及副产物生成的影响;结合催化剂电子性质分析,探究不同金属活性中心对该反应催化性能调控的微观本质原因。
1 计算过程
1.1 计算方法
密度泛函理论(DFT)计算通过Materials Studio 8.0 软件包中Dmol3模块[28-29]完成,计算采用广义梯度近似(GGA)下的交换关联函数(PBE),即GGA-PBE[30-31]。非金属原子采用全电子基组,金属原子采用有效核电势(ECP)[32-33]方法。价电子波函数采用双数值加极化函数(DNP)[34]展开;计算色散校正(DFT-D)考虑范德华作用力[35]。能量、受力和位移收敛标准分别是2.63×10-3kJ/mol,2.63 kJ/(mol·Å)和5×10-3Å。在所有构型优化中,拖尾效应(Smearing)设置为13.13 kJ/mol,其布里渊区积分Monkhorst-Pack 网格参数设置为3×3×1。过渡态计算采用完全线性同步/二次线性同步方法(Complete LST/QST)[36-37],并采用过渡态确认(TS Confirmation)和频率分析(Vibrational Analysis)验证过渡态。所有物种的吸附以及基元反应过程的能量在反应温度(525 K)下进行了吉布斯自由能校正。
采用从头算分子动力学模拟(AIMD)方法[38]评估负载型双原子催化剂的热力学稳定性。在正则系综(NVT)中,时间步长设为1 fs,模拟时间设定为10 ps,并使用速度调节法控温[39]。模拟过程中不限制催化剂体系的对称性。
1.2 催化剂模型
基于已有的实验[40-41]和理论[42-44]研究,催化剂模型构建方法如下:首先构建六角超晶胞大小为p(6×6)的单层石墨烯;去除4 个碳原子,形成双空缺石墨烯;然后,将空缺周围的6 个碳原子用氮原子取代;随后,把金属双原子放在空缺处;最后,通过几何优化获得氮掺杂石墨烯负载金属双原子催化剂的稳定构型(M1M2@NC,其中M1和M2为金属),几何优化过程中活性中心保持弛豫。该结构包括62 个碳原子,6 个氮原子和2 个过渡金属原子,如图1 所示。对于过渡金属原子,考虑了合成气合成C2氧化物普遍采用的3 种金属原子Cu,Rh 和Co。为了避免周期性模型层与层之间的相互作用,真空层厚度设置为15 Å。
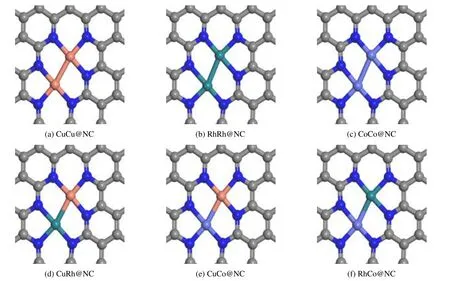
图1 氮掺杂石墨烯负载双原子催化剂的表面结构Fig.1 Surface structure of N-doped graphene supported diatomic catalysts blue ball:N atom;gray ball:C atom;green ball:Rh atom;light blue ball:Co atom;orange ball:Cu atom
2 结果与讨论
2.1 催化剂稳定性
氮掺杂石墨烯负载金属双原子催化剂稳定性可以通过金属活性组分和载体之间的平均结合能与对应体相金属的平均内聚能差值来评价。当差值小于0 时,表明催化剂稳定,且负值越小越稳定,即金属原子不可能在表面聚集成团簇或纳米颗粒。负载型金属双原子催化剂中金属活性组分和载体之间的平均结合能(EaverageBE)以及体相金属的平均内聚能(Eaveragecoh)计算[44]如下:
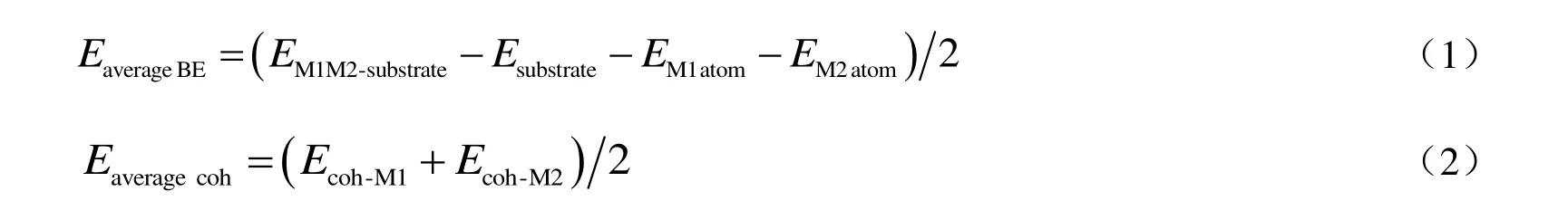
式中:EM1M2-substrate,Esubstrate,EM1atom和EM2atom分别为双原子催化剂结构、氮掺杂石墨烯载体和单原子金属M1和M2的能量,eV;Ecoh-M1和Ecoh-M2分别是体相金属M1和M2的内聚能,eV。
根据公式(1)计算获得CuCu@NC,RhRh@NC,CoCo@NC,CuRh@NC,RhCo@NC 和CuCo@NC的平均结合能分别为-5.57,-7.19,-6.88,-6.84,-7.17 和-6.74 eV;根据公式(2)计算获得CuCu@NC,RhRh@NC,CoCo@NC,CuRh@NC,RhCo@NC 和CuCo@NC 的平均内聚能分别为-3.49,-5.75,-4.39,-4.62,-5.07 和-3.94 eV。这6 类催化剂的平均结合能与平均内聚能的差值分别为-2.08,-1.44,-2.49,-2.22,-2.10 和-2.80 eV。因此,构建的6 类氮掺杂石墨烯负载双原子催化剂均具有较高的稳定性。
2.2 CO 活化生成CHx 中间体
表1 列出了525 K 下合成气制C2氧化物过程中所考虑基元反应的活化自由能(Ga)和反应自由能(ΔG)。

表1 525 K 下不同催化剂上涉及相关基元反应的活化自由能和反应自由能Table 1 Activation energy and reaction energy of related elementary reactions over the catalyst at 525 K
2.2.1 CO 起始活化
CO 起始活化主要存在两种机理:CO 直接解离生成C+O 和CO 加氢生成CHO/COH。如表1 所列,从基元反应的动力学和热力学上看,6 类催化剂上CO 直接解离反应比CO 加氢反应更难发生;同理,CO 加氢更容易生成CHO,而不是COH。另一方面,CuCu@NC,RhRh@NC,CoCo@NC,CuRh@NC,RhCo@NC 和CuCo@NC 催化剂上CHO 生成能垒分别为81.4,110.0,110.5,53.3,94.9和36.6 kJ/mol,表明异核双原子催化剂在动力学上比其对应金属同核双原子催化剂更易于生成CHO,归因于不同金属原子间的协同作用强于同种金属原子。
2.2.2 CH 中间体生成
CH 主要通过CHO 和CHOH 中间体中C—O 键断裂生成,存在2 条路径:
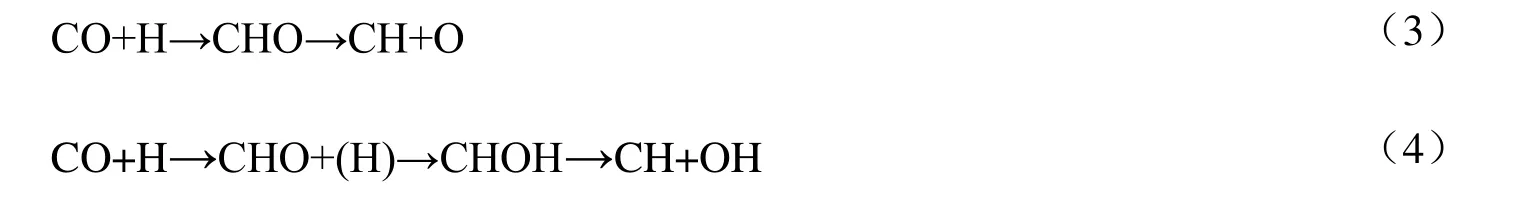
图2 为生成CH 反应势能图,图中TS1~TS6 分别对应表1 中基元反应1~6 的过渡态。由图2可知,在CuCu@NC,RhRh@NC,CoCo@NC,CuRh@NC,RhCo@NC 和CuCo@NC 上,根据式(3)路径生成CH 总能垒分别为530.9,424.9,475.9,537.1,480.1 和542.5 kJ/mol;根据式(4)路径生成CH 总能垒分别为352.8,282.0,359.6,358.1,331.3 和380.1 kJ/mol,从动力学上看,按式(4)路径更易生成CH,因此,该路径为生成CH 的主路径。
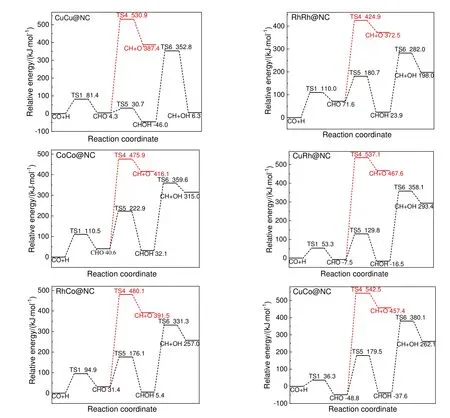
图2 CH 生成反应势能Fig.2 The potential energy profile of CH formation
2.2.3 CH2中间体生成
CH2主要通过CH2O 和CH2OH 中间体中C—O 键断裂生成,存在3 条路径:

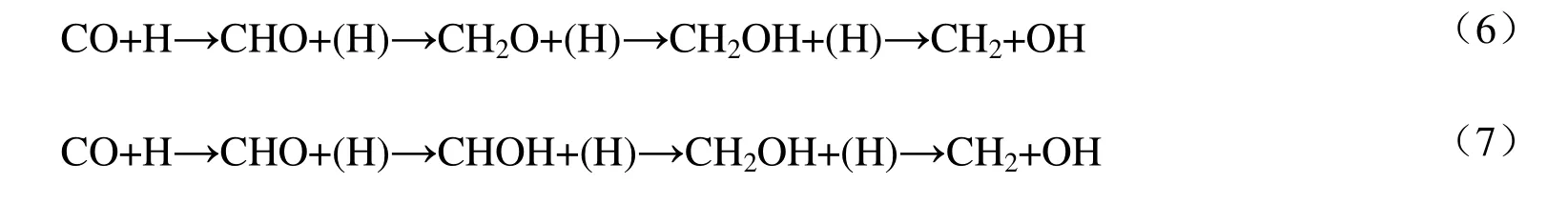
在式(5)路径和式(6)路径中,需要比较CH2O 脱附与其加氢生成CH2OH 及C—O 键断裂反应的难易。DFT 计算获得CuCu@NC,RhRh@NC,CoCo@NC,CuRh@NC,RhCo@NC 和CuCo@NC上CH2O 脱附能分别为4.3,110.8,65.0,24.5,74.3 和-14.0 kJ/mol;CH2O 加氢生成CH2OH 的反应能垒分别为105.0,95.4,113.7,145.5,131.7 和94.0 kJ/mol(见表1);CH2O 解离生成CH2+O 的反应能垒分别为398.6,376.7,285.6,349.3,343.8 和329.3 kJ/mol(见表1)。因此,比较CH2O 脱附能与其加氢和解离反应能垒可知,在CuCu@NC,CoCo@NC,CuRh@NC,RhCo@NC 和CuCo@NC催化剂上,CH2O 倾向于脱附而不参与后续加氢或解离反应,即这5 类催化剂上生成CH2按照式(7)路径进行;在RhRh@NC 上,CH2O 仅参与加氢,而不参与解离反应,即CH2O 通过式(6)路径和式(7)路径生成CH2。图3 为生成CH2反应势能图。由图3 可知,6 类催化剂上CH2生成的最有利路径均为式(7)路径,总能垒分别为184.9,246.9,222.9,129.8,176.1 和179.5 kJ/mol。
图3 CH2 生成反应势能Fig.3 The potential energy profile of CH2 formation
2.2.4 CH3中间体和甲醇生成
CH3仅能通过CH3O 中间体中C—O 键断裂生成,仅存在唯一路径:
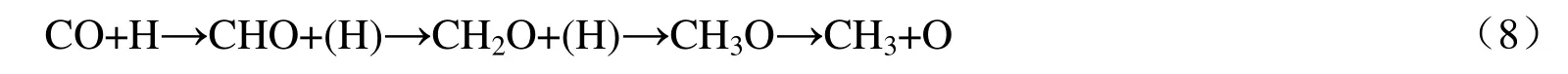
在CuCu@NC,RhRh@NC,CoCo@NC,CuRh@NC,RhCo@NC 和CuCo@NC 上CH2O 加氢生成CH3O 反应能垒分别为60.6,100.9,181.6,100.7,118.3 和146.6 kJ/mol(见表1),比较CH2O脱附能与其加氢生成CH3O 反应能垒大小可得,在RhRh@NC 上CH2O 可以加氢生成CH3O,其余5类催化剂上CH2O 均优先脱附,而不参与其相关反应。因此,仅在RhRh@NC 上能够按照式(8)路径生成CH3,总能垒为193.3 kJ/mol(如图4 所示),其余5 类催化剂均不能生成CH3中间体。
图4 CH3 和CH3OH 生成反应势能Fig.4 The potential energy profile of CH3 and CH3OH formation
而对于CH3OH 生成,存在3 条可能的路径:
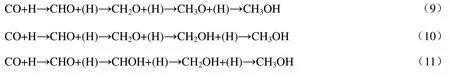
在式(9)路径和式(10)路径中,比较CuCu@NC,RhRh@NC,CoCo@NC,CuRh@NC,RhCo@NC和CuCo@NC 上CH2O 脱附能与其加氢生成CH3O(见表1,其值分别为60.6,100.9,181.6,100.7,118.3 和146.6 kJ/mol)和CH2OH 的反应能垒(见表1,其值分别为105.0,95.4,113.7,145.5,131.7和94.0 kJ/mol),发现仅在RhRh@NC 上CH2O 可加氢生成CH3O 和CH2OH,其余5 类催化剂上CH2O优先脱附,不参与其相关反应。说明在RhRh@NC 上按式(9)~式(11)3 条路径生成甲醇,而其余5 类催化剂仅通过式(11)路径生成甲醇。图4 为生成CH3和CH3OH 反应势能图。由图4 可知,在CuCu@NC,RhRh@NC,CoCo@NC,CuRh@NC,RhCo@NC 和CuCo@NC 上甲醇生成的最有利路径为式(11),其总能垒分别为109.5,180.7,222.9,129.8,176.1 和179.5 kJ/mol。
2.2.5 最有利CHx单体和甲醇
图5 为CHx(x为1~3)和CH3OH 生成最有利路径能垒。由图5可知,在CuCu@NC,CoCo@NC,CuRh@NC,RhCo@NC 和CuCo@NC 上最有利CHx中间体为CH2,总能垒分别为184.9,222.9,129.8,176.1 和179.5 kJ/mol;在RhRh@NC 上最有利CHx中间体为CH3,总能垒为193.3 kJ/mol。同时,在CuCu@NC,RhRh@NC,CoCo@NC,CuRh@NC,RhCo@NC 和CuCo@NC 上,生成甲醇最有利路径总能垒分别为109.5,180.7,222.9,129.8,176.1和179.5 kJ/mol。
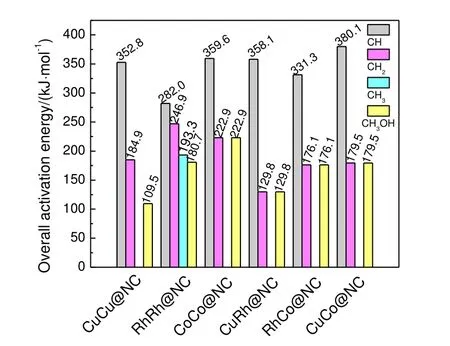
图5 CHx和CH3OH 生成最有利路径能垒Fig.5 The overall barrier for the most favorable routes of CHx and CH3OH formation
图6 为CHx(x为2 和3)和CH3OH 生成的最有利路径势能图。CuCu@NC,CoCo@NC,CuRh@NC,RhCo@NC 和CuCo@NC 上,CH2生成最有利路径均为式(7)路径;CH3OH 生成最有利路径均为式(11)路径,这两条路径都经过共同中间体CH2OH。而RhRh@NC 上,CH3生成最有利路径为式(8)路径,CH3OH 生成最有利路径为式(11)路径,这两条路径都经过共同中间体CHO。故起始于共同中间体,通过最有利单体CHx(x为2 和3)和CH3OH 生成反应的能垒差可以定量评价CHx选择性,差值越负,CHx选择性越高,即CHx选择性顺序为RhCo@NC(-113.7 kJ/mol)>CoCo@NC(-50.8 kJ/mol)>RhRh@NC(12.6 kJ/mol)>CuCo@NC(105.3 kJ/mol)>CuRh@NC(126.6 kJ/mol)>CuCu@NC(241.0 kJ/mol)。因此,RhCo@NC 和CoCo@NC 催化剂利于CH2生成,能够有效抑制甲醇形成,提高CH2的选择性。
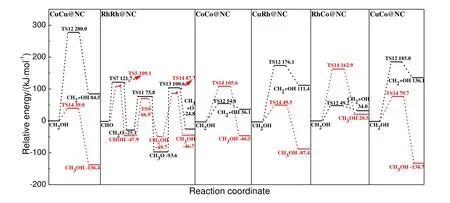
图6 生成CHx(x 为2 和3)和CH3OH 最有利路径势能Fig.6 The potential energy profile for the most favorable routes of CHx (x=2-3) and CH3OH
2.2.6 CHx中间体和甲醇生成微观动力学
微观动力学计算能够考虑反应温度、压力以及反应中间体覆盖度等对反应性能的影响,故进行了典型实验条件下(PCO为400 kPa,PH2为800 kPa,T为525 K)合成气制CHx反应的微观动力学计算,计算方法与先前研究一致[45]。表2 给出CHx生成过程中物种覆盖度、CHx和CH3OH 生成速率及其选择性。
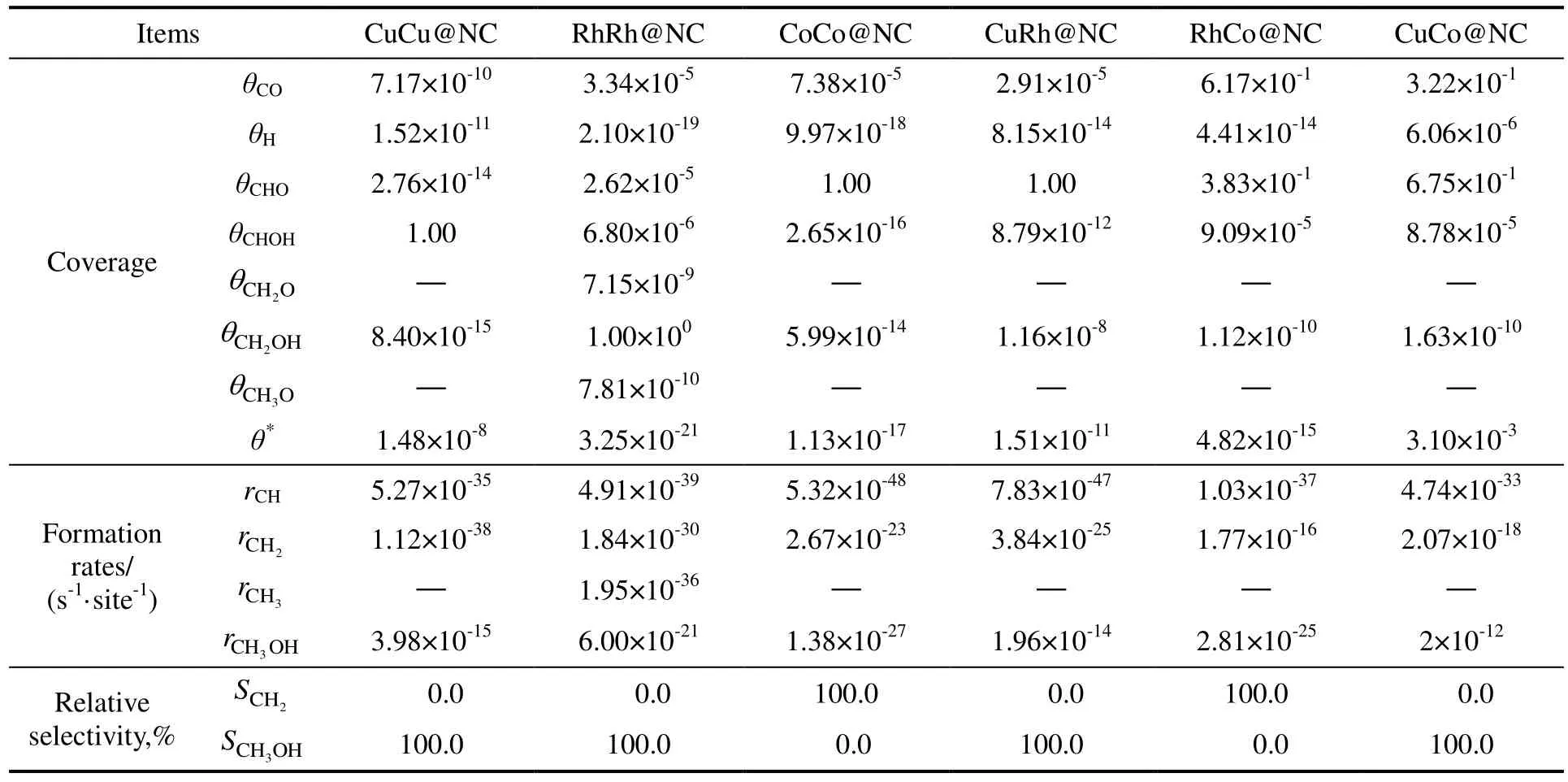
表2 525 K 下催化剂上反应物种的表面覆盖度、CHx 和CH3OH 生成速率及选择性Table 2 The surface coverage of all species,the formation rates and selectivity of CHx and CH3OH over the catalyst at 525 K
在CoCo@NC,CuRh@NC,CuCo@NC 和RhCo@NC 上,CH2生成速率远快于CH 和CH3,故CH2是这4 类催化剂上最有利CHx单体,与DFT 计算结果一致。在CuCu@NC 和RhRh@NC 上CH和CH2生成速率最快,与DFT 计算结果不一致,这是由于CHx生成速率不仅取决于速率常数,还取决于反应物种覆盖度。微观动力学表明:CuCu@NC,RhRh@NC,CuRh@NC 和CuCo@NC 对生成CH3OH 具有较高选择性;但是,CoCo@NC 和RhCo@NC 可以抑制甲醇生成,对生成CH2具有较高选择性。
2.3 C2 氧化物生成
合成气转化生成CHx单体后,CHx可能发生4 类反应:(1)CHx解离;(2)CHx加氢生成CH4;(3)CO/CHO 插入CHx生成C2氧化物;(4)CHx自耦合生成C2烃类。甲烷和C2烃类的生成会降低C2氧化物选择性。DFT 计算结果表明,在CuCu@NC,CoCo@NC,CuRh@NC,CuCo@NC 和RhCo@NC上最有利CHx单体均为CH2,故CH2作为碳链生成单体,考虑了CH2相关反应;在RhRh@NC 上最有利CHx单体为CH3,故CH3作为碳链生成单体,考虑了CH3相关反应。
图7 给出了6 类催化剂上有利单体相关反应的总能垒图。在RhRh@NC 上,生成CH2,CH4,C2H6,CH3CO 和CH3CHO 反应的总能垒分别为31.9,83.5,286.2,193.4 和170.2 kJ/mol,即C2氧化物生成困难;在CuCo@NC 上,不利于C2氧化物生成。故这2 类催化剂不易生成C2氧化物。而在CuCu@NC,CoCo@NC,CuRh@NC 和RhCo@NC 上,通过CHO 和CH2反应生成C2氧化物最有利,其总能垒分别为109.7,29.4,51.5 和40.2 kJ/mol,故这4 类催化剂能够生成C2氧化物,其中CoCo@NC,CuRh@NC和RhCo@NC 催化剂呈现出高活性。综上所述,无论是同核催化剂还是异核催化剂,生成CH 均比生成CH2和CH3困难,即6 类催化剂都不利于CH 生成。在CuCu@NC,CoCo@NC,CuRh@NC,RhCo@NC 和CuCo@NC 上,最有利单体为CH2,且CH2生成在异核催化剂上容易于同核催化剂;在RhRh@NC 上,最有利单体为CH3。同时,6 类催化剂中只有RhCo@NC 和CoCo@NC 催化剂可以抑制甲醇生成,其余4 类催化剂均是甲醇生成优于CHx生成。进一步,RhCo@NC 上CH2生成活性远大于CoCo@NC 催化剂。因此,筛选出RhCo@NC 催化剂能够高活性高选择性地生成CH2,其后CHO 插入CH2能够高活性且高选择性生成C2氧化物。
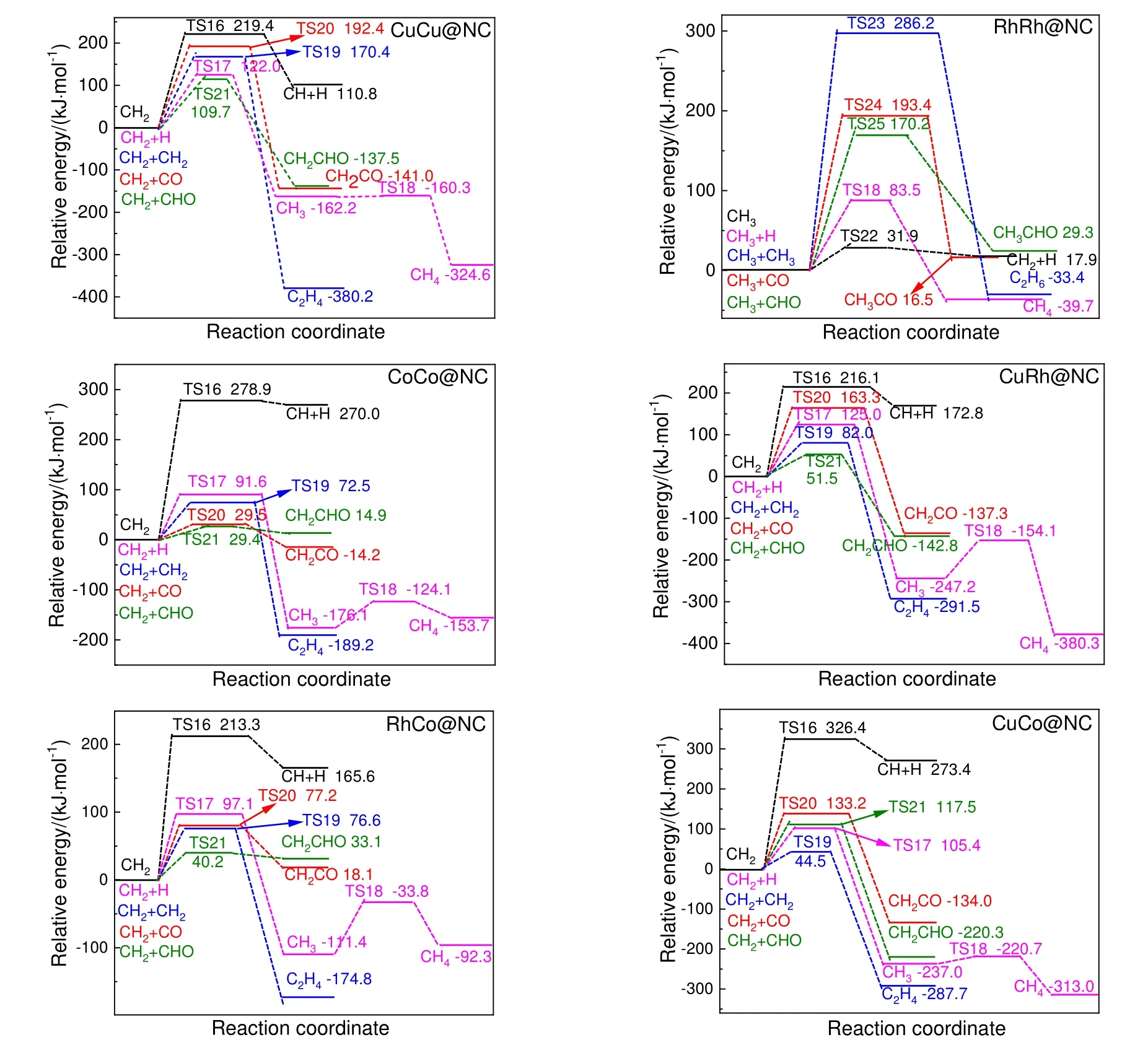
图7 CHx(x 为2 和3)相关反应势能Fig.7 The potential energy profile of the reactions related to CHx (x=2,3)
2.4 电子性质分析
负载型金属双原子催化剂的催化性能与其电子性质有着密不可分的联系[46]。如图8 所示,CuCu@NC,RhRh@NC 和CoCo@NC 同核催化剂的电荷分布对称,但CuRh@NC,RhCo@NC 和CuCo@NC 异核催化剂的电荷分布不对称,由于电荷分布不对称导致异核双原子催化剂活性比同核双原子催化剂高。差分电荷密度表明金属双原子将电子转移到氮原子,正负电荷主要分布在金属双原子和氮原子之间。Bader 电荷表明CuCu@NC,RhRh@NC,CoCo@NC,CuRh@NC,RhCo@NC 和CuCo@NC 中金属双原子电荷数分别为0.681,0.534,0.682,0.567,0.606 和0.665 e。
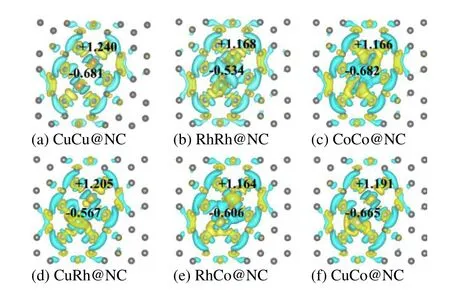
图8 催化剂表面电荷差分密度Fig.8 Differential charge density of catalyst surface
催化剂表面电荷转移量与CHx生成活性关系如图9 所示,当电荷转移量在0.552~0.671 e 时,催化活性较好,其归因于异核双原子与氮掺杂石墨烯电荷转移量居于同核双原子中间,使得不同金属活性组分表现出协同效应。
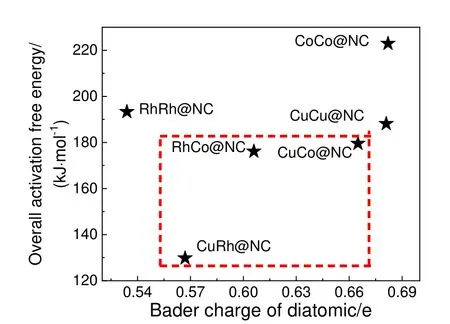
图9 金属双原子Bader 电荷与CHx生成活性关系Fig.9 The relationship between Bader charge of diatomic metals and the activity of CHx formation
2.5 催化剂热稳定性评价
由于RhCo@NC 催化剂在合成气合成C2氧化物反应中具有高活性和高选择性,进一步采用AIMD 模拟评估实验条件下该催化剂的热稳定性,结果如图10。如图10 所示,温度和能量在平衡态附近振荡,在525 K高温下,金属双原子RhCo 与其相邻的6 个氮原子仍能紧密结合,且RhCo@NC 催化剂几何结构无明显变形,表现出较高的热力学稳定性[44]。目前,实验已经能够制备氮掺杂石墨烯负载金属双原子催化剂[41],所以RhCo@NC 催化剂在合成气制C2氧化物反应中具有潜在的应用前景。
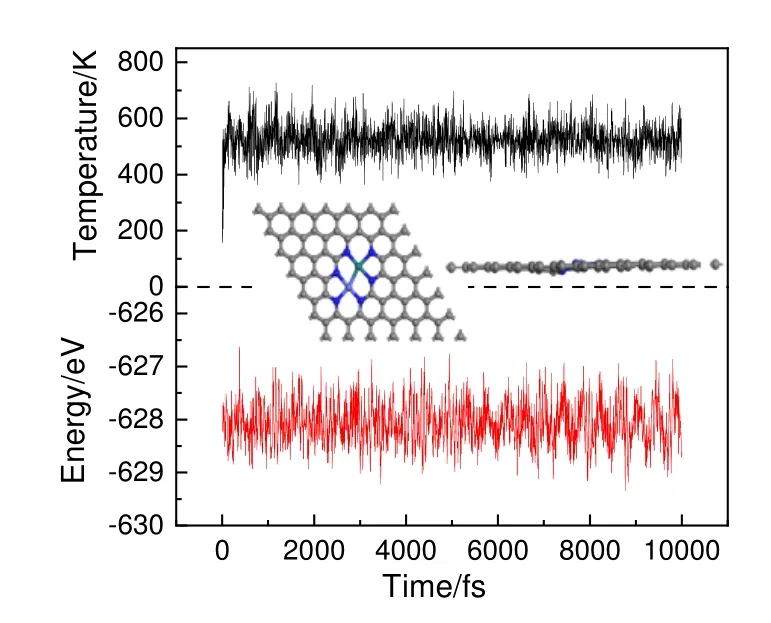
图10 RhCo@NC 催化剂的AIMD 模拟Fig.10 AIMD simulation of RhCo@NC catalyst
3 结论
采用密度泛函理论计算,研究了氮掺杂石墨烯负载不同金属双原子催化剂上合成气合成C2氧化物的反应性能,研究结果表明负载双原子催化剂中异核相邻原子间协同效应提高了合成气合成C2氧化物的活性和选择性。在合成气转化生成CHx过程中,异核双原子催化剂活性比同核双原子催化剂高,即不同金属间的协同效应比同种金属更好,促进了关键中间体CH2生成。一旦关键中间体CH2生成,其后CHO 与CH2反应能够高活性和高选择性地生成C2氧化物。在6 类催化剂中,RhCo@NC 催化剂是合成气合成C2氧化物最具潜力的催化剂,具备高活性和选择性以及高的热稳定性。电子性质分析进一步表明,RhCo 金属活性中心通过优化电荷分布,从而提高催化性能。
