UV/H2O2对化工废水中2,6-二氯吡啶的降解研究
2021-03-05谷振超邬东陈莉荣
谷振超,邬东,陈莉荣
(内蒙古科技大学能源与环境学院,内蒙古包头,014010)
2,6-二氯吡啶(2,6-DCLPY)是一种应用广泛的含氮杂环化合物,不仅是许多杀菌剂、香料和特定农医药物的基本原料[1],还经常作为染料、树脂和部分精细化学品生产过程中的溶剂使用[2].因此,它普遍存在于焦化废水、医药废水和精细化工等工业废水中[3-7].这些化工废水中的2,6-DCLPY 因结构中含有难降解的吡啶环结构并具有较强的生物毒性,难以被一般物化处理工艺或生化手段去除[8-9].此外,它具有较强的水溶性和生物活性,可以在土壤、水体和沉积物中迁移和富集,对生态安全构成直接威胁[10].已经从包括北海(0.07 ng/L)、英国沿海(0.13 ng/L)、易北河(8.76 ng/L)等环境水体中检测出2,6-DCLPY及多种芳香族衍生物的存在[11].因此,如何高效去除这类含吡啶环结构的污染物成为了水污染领域的研究热点.但由于取代基团的影响,这些化合物与目前研究者们普遍关注的典型污染物之间存在一些性质上的差别.比如2,6-DCLPY 与吡啶相比,就具有碱性弱难以质子化、水溶性差难极化、环上电子密度低不易发生亲电反应的特点,探明这类化合物的降解特性是对现有研究的重要补充.
目前为止,通过生物降解[8]、光化学[12]和高级氧化[13-15]等方法去除这类物质的研究均有报道,而高级氧化技术成为当前的主流研究方向[16-18].在多种生成·OH 的体系中,紫外光活化H2O2由于具备条件易控、产·OH 效率高、较低的消光系数(ε254nm=19.6 L·mol-1·cm-1)等特点而受到关注.相较于电催化、金属离子催化等手段,通过UV 活化H2O2的过程受环境因素影响较小;无需引入新物质,可以避免催化剂分离的问题;且生成紫外光的技术已相对成熟.已有研究表明该体系可以实现系列2-卤代吡啶的高效光解除去和矿化,对2-氯代吡啶的去除率高达95%[19].
基于此,本文选择2,6-二氯吡啶为主要研究对象,设计了其在UV/H2O2体系下的降解实验,研究pH、H2O2用量、底物初始浓度等因素对2,6-DCLPY降解和矿化程度的影响;考察4 种化工废水中含量较高的阴离子在高浓度条件下对降解过程的影响;并分析了吡啶环上不同取代基对降解程度和速率的影响,以期为实际工程中采用UV/H2O2工艺去除氯代吡啶类氮化物提供参考.
1 材料与方法
1.1 试剂与仪器
实验用水:自配2,6-二氯吡啶(2,6-DCLPY)模拟废水,将1 g 的2,6-二氯吡啶溶解于甲醇中并定容至100 mL,常温下保存不超过24 h.实验时取0.2、0.4、0.6 mL 配置好的2,6-DCLPY 溶液加入到200 mL 去离子水中,得到底物质量浓度为10、20、30 mg/L 的2,6-DCLPY 溶液.
试剂与仪器:2,6-二氯吡啶(97%,阿拉丁);过氧化氢30%(分析纯,天津市风船化学试剂科技有限公司);碳酸氢钠(NaHCO3),硝酸钠(NaNO3),氯化钠(NaCl)和硫酸钠(Na2SO4)(均为分析纯,天津市风船化学试剂科技有限公司);精密酸度计(PHS-3C,上海雷磁仪器厂);紫外可见分光光度计(UV180G,天津光泽科技有限公司);TOC 分析仪(TOC-VCPN,日本岛津公司);超纯水机(GWA-UN1-20,北京普析通用仪器有限公司);COD 快速测定仪(5B-3C,连华科技);光化学反应器(ZQ-GHX-I,上海争巧科学仪器有限公司,反应暗箱配备高压汞灯光源,波长250~720 nm,功率300 W,工作电压(110 ± 10)V,汞灯置于双层全石英冷阱内,反应容积为250 mL,反应器中心光强Is=1.72 mW·cm-2).
1.2 实验装置与方法
UV-H2O2反应装置如图1 所示,采用连续反应方式.反应器总体积为2.0 L,有效反应容积250 mL,高为340 mm,外筒直径为90 mm.石英冷阱高为375 mm,外径60 mm,内径40 mm.直径为20 mm 的300 W 紫外高压汞灯(厚1.8 mm 的石英套管保护灯管)竖直放置于内筒中央,连接循环冷却水保证反应体系维持在室温,反应器放置于黑箱中.首先用H2SO4与NaOH 调节水样至所需pH 值,然后将200 mL 水样从其中一个取样口部倒入反应器中,投加适量30%H2O2,插入紫外灯管并固定,开启冷凝循环水和风扇,然后接通电源开启紫外灯和磁力搅拌器(搅拌速度80 r/min),反应开始进行.通过基础实验确定最优反应条件,在最优反应条件下,延长反应时间,定时取样,测定水样特征吸光度表征水样中2,6-二氯吡啶的降解程度,测定TOC 值表征水中有机物的矿化程度.

图1 UV-H2O2反应装置示意Fig.1 Schematic diagram of UV-H2O2reaction device
1.3 分析方法
pH 值用pH 计测量;总有机碳采用TOC 检测仪,水样经0.45 μm 滤膜过滤后进行TOC 的测定;采用紫外可见分光光度法(UV180G,石英比色皿为1 cm)测得2,6-二氯吡啶最大吸收峰(271 nm),然后用标准曲线法测定降解过程中2,6-二氯吡啶的质量浓度,检测范围为2~32 mg/L,所有测定都是将样品调至pH 为7(±0.2)下进行;实际水样的总碱度用国标法测定;实际水样的COD 用连华科技COD 快速测定仪(5B-3C)测定.
2 结果与讨论
2.1 2,6-二氯吡啶的降解
为了更好地评估UV/H2O2系统下2,6-DCLPY的降解效果,建立伪一级动力学模型(方程1)计算2,6-DCLPY 降解的结果.建立量子产率(φ2.6-DCLPY)模型(方程2)来计算UV 的量子产率进而评估UV 的直接光解效果.


式(1)中:[2,6-DCLPY]0是2,6-二氯吡啶的初始质量浓度(mg/L),[2,6-DCLPY]是2,6-二氯吡啶在t时刻的质量浓度(mg/L),kobs是伪一级动力学反应速率常数(min-1).
式(2)中:Uλ是在特定紫外辐射波长下1 mol 光子(J Einstein-1)的能量(Uλ=hcNA/λ,得271 nm 下的单位摩尔光子能量为441 326.49 J Einstein-1),Is是平均光照强度,这里是17.2 Wm-2,ε2,6-DCLPY由朗伯-比耳定律计算(A=ε2,6-DCLPYlc).
由图2(a)可见,当2,6-DCLPY 的初始质量浓度为10 mg/L,H2O2浓度为2.5 mmol/L,未调节配置模拟废水的pH 值(pH 值接近中性),反应120 min后,单独的H2O2氧化技术对2,6-DCLPY 的最大降解率仅为10%左右,反应速率常数为0.008 min-1.相同实验条件下采用UV 系统降解2,6-DCLPY 的最大降解率为48.8%,降解速率也同步提高了5 倍.出现这样的结果可能是因2,6-DCLPY 的最大吸光带在271 nm,此波长下的计算φ2.6-DCLPY为0.031,这虽与之前的研究相比略低[20],但是这与高压汞灯的主波长313 nm 较为接近,因此基本保证了用于降解2,6-DCLPY 的光量子充足,故而底物可在紫外光照射下发生直接光解.作为对照,UV/H2O2的联合显著提高了反应体系对2,6-DCLPY 的降解程度(最大降解率77.3%,反应速率常数0.064 min-1),这可能是由于UV/H2O2体系下2,6-DCLPY 除了直接光解的途径外,还会被紫外光激发产生的·OH 氧化降解,见式(3)(4).

为确定是否存在·OH 参与2,6-DCLPY 降解过程,向溶液中添加对·OH 具有强淬灭作用的叔丁醇(TBA)进行对比实验[21].在不同pH 条件下将200 mmol·L-1的TBA 加入到原反应体系进行降解实验,发现2,6-DCLPY 的降解均被明显抑制.图2(b)显示了使用淬灭剂后的实验结果,在没有添加任何淬灭剂的情况下最优可除去77.3%的2,6-DCLPY,而TBA 的添加使得在pH 值为3、7.2、9 的条件下2,6-DCLPY 的降解率分别降低至39.8%,32.2%和27.3%.
自由基抑制实验表明:不含α-H 的叔丁醇在中性及弱碱性条件下对羟基自由基的清除效果明显(kobst=3.8-7.6×108L·mol-1·s-1)[22-23].结合空白组作为对照可知,在酸性、中性和碱性条件下羟基自由基都有产生,但在中性及弱碱性条件下羟基自由基的活性更强.进一步地通过方程(1 -(kobs,TBA/kobs))来评估·OH 对2,6-DCLPY 降解的贡献率,得出UV/H2O2体系下由·OH 氧化引起的2,6-DCLPY 去除率占总去除率的74.1%,而起协同作用的UV 只提供了25.9%的降解贡献率.因此,·OH 是UV/H2O2体系中降解2,6-DCLPY 的主要活性物质.

图2 氧化体系及不同pH 下自由基清除剂对2,6-DCLPY 降解的影响Fig.2 Effect of oxidation system and scavenger at different pH on 2,6-DLPY degradation
2.2 初始H2O2浓度对降解的影响
如图2 所示,UV 体系下H2O2的投加能够加快2,6-DCLPY 的去除,且在一定范围内随H2O2浓度升高,2,6-DCLPY 的降解效率和反应速率越高.因此,UV/H2O2对2,6-DCLPY 的降解符合准一级反应动力学,不同H2O2投加量时2,6-DCLPY 反应速率常数Kobs见图3 内插图.当H2O2浓度小于2.5 mmol/L,2,6-DCLPY 降解速率随着H2O2用量的增加增长较快,但当H2O2用量超过2.5 mmol/L 时,2,6-DCLPY降解速率随着H2O2浓度的提高缓慢下降.由于H2O2可以与·OH 发生反应,见式(6),因此,过量的H2O2可能会和目标污染物竞争·OH 而影响其与底物的反应,同时高H2O2投加量下可能会存在·OH 的自清除和自复合,见式(5)(6)[24].据此确定H2O2和2,6-DCLPY 的最佳摩尔比为37 ∶1.

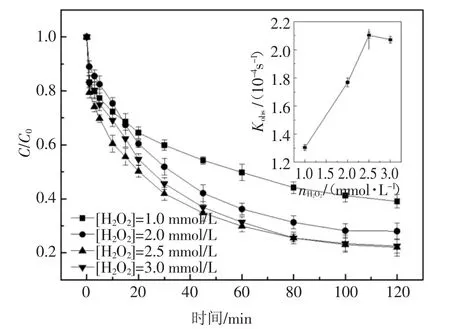
图3 H2O2浓度对2,6-DCLPY 降解的影响Fig.3 Effect of H2O2concentration on 2,6-DCLPY degradation
2.3 2,6-二氯吡啶初始浓度对降解的影响
在过氧化氢与2,6-DCLPY 摩尔比相同的条件下,以初始质量浓度10、20、30 mg/L 的2,6-DCLPY溶液为研究对象,考察UV/H2O2体系对不同浓度2,6-DCLPY 的降解效果及矿化程度的影响.由图4可知,2,6-DCLPY 降解率和TOC 去除率都随反应时间延长逐渐增大.反应80 min 时,初始质量浓度10、20、30 mg/L 的溶液中2,6-DCLPY 去除率分别为73.82%、71.33%、66.77%;但溶液中有机物的矿化程度较低,在降解120 min 后TOC 去除率均低于50%.较低的矿化程度是多种原因所致,首先2,6-DCLPY 中吡啶环受到多氯取代的影响,电子密度明显下降,增大·OH 氧化开环难度;反应前后可以观察到pH 值的明显变化,说明在中间过程Cl 原子转化为氯化氢,较低的pH 会抑制后续降解过程;N 原子转变为小分子有机胺后难以进一步矿化;随着反应时间延长,初始添加的H2O2可能不足以提供充足的·OH.
表1 为3 种反应动力学拟合方程及参数,从中可以看出10、20、30 mg/L 时2,6-DCLPY 的反应表观速率常数分别为1.481、1.457、1.276,2,6-DCLPY的降解速率随初始浓度的升高而下降.整个降解过程的TOC 去除率增长缓慢,低底物质量浓度(10 mg/L)的TOC 最终去除率约为50%,高底物质量浓度(30 mg/L)的TOC 去除率仅为26.5 %,这表明大部分2,6-DCLPY 转化为中间产物后难以进一步矿化.

图4 底物质量浓度对2,6-DCLPY 降解的影响Fig.4 Effect of substrate concentration on 2,6-DCLPY degradation

表1 UV/H2O2降解不同初始质量浓度2,6-DCLPY溶液的动力学及TOC 参数Tab.1 Kinetics and TOC parameters of different initial concentrations on 2,6-DCLPY degradation by UV/H2O2
2.4 pH 值对降解的影响
实验前用0.1 mol/L 的HCl 和NaOH 调节体系的初始pH 值分别为3、5、7.2(2,6-DCLPY 原液pH 值为7.2,误差为±0.02)、9、11 进行降解实验,结果见图5.

图5 pH 值对2,6-DCLPY 降解的影响Fig.5 Influence of pH on 2,6-DCLPY degradation
结果显示酸性条件下降解程度总体较低,酸性增强时降解率降低,但降解速度在实验初期会出现反转.这可能是由于弱酸性(pH=5)条件下H2O2较稳定,而较强酸性(pH=3)条件下初期高浓度的H2O2容易与·OH 反应产生HO2·,从而暂时降低了·OH 的浓度,使降解速率下降[25-26],见式(7);当溶液呈中性和弱碱性时2,6-DCLPY 降解程度较高,同时kobs和降解速率在pH 值5~9 范围内随pH 值提高而增加,当pH 在7~9 的范围内时反应速率常数均在0.011 min-1以上.
考虑到降解2,6-DCLPY 的主要活性物质是·OH,而H2O2碱性电离产生的,以及OH-都会与其发生反应,见式(8)[27]、式(9)[28],从而抑制降解过程,因此理论上随pH 上升该体系对2,6-DCLPY 的降解能力会逐渐下降,但实验结果显示降解率在中性(pH 7.2)时达到最高.这可能是受2,6-DCLPY 的形态变化影响,在中性及弱碱性条件下2,6-DCLPY为分子态,而较低pH 值下会结合质子变为阳离子态,此时其结构中吡啶环的电子密度降低,难以被·OH 氧化,从而导致降解率降低,实验中碱性条件下降解反应的二级速率常数大于酸性条件也支持该解释.

2.5 水相中无机离子的影响
化工浓盐水中常见无机阴离子主要为Cl-,,因此设计实验研究以上离子对2,6-DCLPY 降解过程的影响.由于Cl-和通常含量较高,是主要的盐度组成,而,含量相对较低,因此分别选择质量浓度范围0~50 g/L 和0~5 g/L作为研究区间.实验结果如图6(a)(b)(c)(d)所示.
由图6(a)可知,Cl-初始质量浓度从0 增加到10 g/L 时,UV/H2O2体系降解2,6-DCLPY 80 min 后去除率从73.8%下降到66.9%,而当Cl-初始质量浓度进一步增加到50 g/L 时,去除率为44.8%,与Cl-初始浓度为0 时相比,降解率下降29.0%,降解率差异并不巨大.在UV/H2O2体系下,Cl-与·OH 形成ClOH-的逆反应非常迅速,使得Cl-对·OH 的清除作用很弱.产生的ClOH-.进一步反应为Cl·和Cl2·-也有一定的氧化电势(E0(Cl·/Cl-)=2.41 V,E0(Cl2·-/2Cl-)=2.41 V)[29],如式(10)(11)(12)中所列[29-30].因此,理论上Cl-对体系中·OH 的浓度几乎没有影响,不会显著影响2,6-DCLPY 的降解.实验中Cl-对2,6-DCLPY 降解的轻度抑制可能是由于高盐度下参与竞争·OH 的Cl-数目巨大而阻碍了底物和·OH 的结合.
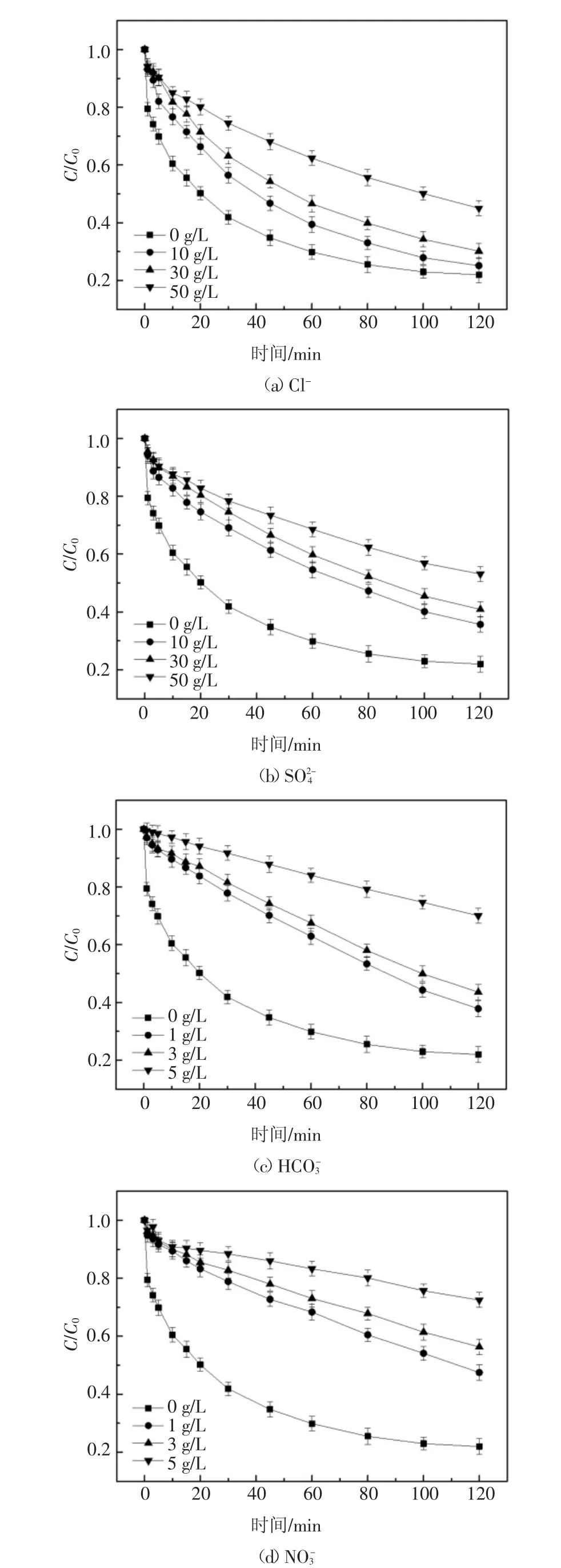
图6 无机阴离子对2,6-DCLPY 去除的影响Fig.6 Effect of inorganic anions on 2,6-DCLPY degradation
2.6 吡啶环上不同取代基对降解效果的影响
从表2 中可以看出,吡啶环上存在强给电子基团,3-氨基-2-氯吡啶的降解速率表现为零级反应,B.F.Abramovic 等[38]对其进行了Langmuir-Hinshelwood(L-H)的动力学模型拟合分析,计算得出吸附系数为1.2×104dm3/mol.这说明反应物极为活泼,在催化剂表面吸附的瞬间即被分解,因此反应速率与反应物浓度无关,只与催化剂的反应位点数量相关.最终3-氨基-2-氯吡啶的降解率为95.0%,TOC 去除率也达90.0%.
而吡啶、2-氯吡啶、2,6-二氯吡啶在各自反应体系下均为准一级反应,三者的反应速率表现为吡啶>2-氯吡啶>2,6-二氯吡啶,这应该是由于吸电子基团Cl 的影响.根据文献报道,吡啶环降解的主要途径都要通过·OH 对其进行亲电取代反应以生成中间产物[41],而氯取代基使得吡啶环上电子密度下降,降低了吡啶环的反应活性,抑制了降解的进行,氯取代基团越多则该影响越显著.因此吡啶环上电子密度最低的2,6-二氯吡啶降解和矿化程度最低.

表2 不同取代吡啶的降解效果Tab.2 Degradation effect of different substituted pyridines
2.7 2,6-二氯吡啶在实际水体中的降解
为研究UV/H2O2对实际水体中2,6-DCLPY 的去除效果,取包头某焦化企业的RO 浓水作实际水样(水质参数如表3),向其中加入10 mg/L 2,6-DCLPY 和3.5 mmol/L 的H2O2进行降解实验.实验结果如图7 所示,与纯水中2,6-DCLPY 的降解相比,2,6-DCLPY 在实际水体中的降解受到抑制.考虑到所取实际废水含盐量高,其中主要阴离子Cl-和都会抑制降解过程,如上述讨论,这一实验结果符合预期.

表3 实际水样的水质参数Tab.3 Water quality parameters of actual water samples

图7 2,6-二氯吡啶在实际水体中的降解Fig.7 Degradation of 2,6-dichloropyridine in actual water
3 结论
1)·OH 是UV/H2O2体系中导致2,6-DCLPY 降解的主要活性物质,由其氧化引起的2,6-DCLPY降解贡献了总降解量的74.1 %,而UV 引发的直接光解占另外25.9 %.2,6-DCLPY 的降解过程符合准一级反应动力学方程,2,6-DCLPY 的降解率随H2O2用量的增加而逐渐提高,但是过量的H2O2能够成为·OH 的淬灭剂.
2)实验确定过氧化氢和2,6-DCLPY 的最佳摩尔比为37 ∶1,pH 为7.2 时降解效率达到最高.不同底物浓度的TOC 去除率测定实验表明,大部分2,6-DCLPY 转化为中间产物后难以进一步矿化.
3)UV/H2O2体系中,Cl-和的存在对2,6-DCLPY 的降解均有抑制,其离子浓度越高抑制作用越明显和的抑制效果更加显著,因此2,6-DCLPY 在实际水体中的降解过程相比于纯水中会受到一定程度的抑制.
4)给电子基团如氨基有利于吡啶类污染物的降解,吸电子基团如氯会抑制吡啶类污染物的降解,因此在吡啶、2-氯吡啶、2,6-二氯吡啶和3-氨基-2-氯吡啶四种物质中,电子密度最低的2,6-二氯吡啶最难降解和矿化.
