Neurohumoral, cardiac and inflammatory markers in the evaluation of heart failure severity and progression
2021-03-03EkaterinaPolyakovaEvgenyMikhaylovDmitrySoninYuriCheburkinMikhailGalagudza
Ekaterina A Polyakova, Evgeny N Mikhaylov, Dmitry L Sonin, Yuri V Cheburkin,Mikhail M Galagudza
Almazov National Medical Research Centre, Saint-Petersburg, Russia
ABSTRACT Heart failure is common in adult population, accounting for substantial morbidity and mortality worldwide. The main risk factors for heart failure are coronary artery disease, hypertension, obesity, diabetes mellitus, chronic pulmonary diseases, family history of cardiovascular diseases, cardiotoxic therapy. The main factor associated with poor outcome of these patients is constant progression of heart failure. In the current review we present evidence on the role of established and candidate neurohumoral biomarkers for heart failure progression management and diagnostics. A growing number of biomarkers have been proposed as potentially useful in heart failure patients, but not one of them still resembles the characteristics of the “ideal biomarker.” A single marker will hardly perform well for screening, diagnostic, prognostic, and therapeutic management purposes. Moreover, the pathophysiological and clinical significance of biomarkers may depend on the presentation, stage, and severity of the disease. The authors cover main classification of heart failure phenotypes, based on the measurement of left ventricular ejection fraction, including heart failure with preserved ejection fraction, heart failure with reduced ejection fraction,and the recently proposed category heart failure with mid-range ejection fraction. One could envisage specific sets of biomarker with different performances in heart failure progression with different left ventricular ejection fraction especially as concerns prediction of the future course of the disease and of left ventricular adverse/reverse remodeling. This article is intended to provide an overview of basic and additional mechanisms of heart failure progression will contribute to a more comprehensive knowledge of the disease pathogenesis.
Heart failure (HF) is an epidemic-wide disease that has a significant burden on clinical and public health systems with significant mortality, morbidity, and high hospitalization rate.[1]The lifetime risk of developing HF is approximately one in five for a 40-year-old in Europe and North America.[2]The main risk factors for HF are coronary artery disease (CAD), hypertension, obesity, diabetes mellitus, chronic pulmonary diseases, family history of cardiovascular diseases, cardiotoxic therapy. The main factor associated with poor outcome of these patients is constant progression of HF.[1]
Numerous clinical and experimental studies indicate that HF progression is largely determined by the stable activation of “neurohumoral” systems:sympathetic nervous system (SNS) and renin-angiotensin-aldosterone system (RAAS).[3]One of physiological function of this “neurohumoral” system is a cardiovascular homeostasis, however, SNS hyperactivation and oversecretion of norepinephrine, angiotensin II, and aldosterone are also known to be toxic to the heart. Another marker of HF compensation is the blood level of natriuretic peptide which is elevated in HF patients with preserved systolic function. Other than SNS, RAAS hormones and natriuretic peptides, neurohumoral markers have not been studied properly in patients with HF progression.[4]
Timely diagnostic and treatment slows down the HF progression. Most common therapeutic options include drugs inhibiting the RAAS and SNS activation, as well as device implantation in selected patients. Such interventions decrease the arrhythmic burden and stabilize hemodynamics. There is a growing interest in the optimization of HF management as well as in establishment and validation of novel diagnostics and therapeutic strategies.[4]This leads to investigation and use of biomarkers not only for screening, diagnosis, and risk stratification,but also for early prophylactics and patient-oriented treatment.[2,4,5]
HF diagnostics and management is associated with a number of biomarkers, but only few of them,such as B-type natriuretic peptides, are used routinely in clinical practice. For a biomarker to have real clinical value, it should meet two main criteria: simplicity of the preanalytical stage, economic availability.[6]The ideal biomarker should also be specific and provide information on cardiac involvement in the different HF stages, stratify risk and benefit in therapeutic decision making. Although there is extensive research on biomarkers in HF progression (e.g., galectin 3, soluble suppression of tumorigenicity protein 2, copeptin, adrenomedullin etc.), there is no definite evidence to recommend them for clinical practice. Prevention of HF progression is successful with effective treatment. Table 1 describes the effects of various drugs for HF patients on the level of certain circulating biomarkers.[6,7]
The success of the research regarding potential new HF biomarkers also depends on the methods of mathematical data processing, starting from the mere demonstration of a statistically significant association with a prespecified end-point to in silico modeling.[8-10]The role of nontraditional risk factors such as understudied peptides, hormones and nervous system effects in the pathogenesis and progression of heart failure has yet to be completely investigated. Resulting knowledge and evidence could lead to the development of the most comprehensive “neurohumoral model” of HF and lead to new effective therapeutic strategies.
In the current review we present evidence on the role of established and candidate neurohumoral biomarkers for HF progression management and diagnostics. We cover main classification of HF phenotypes, based on the measurement of left ventricular ejection fraction (LVEF), including HF with preserved LVEF (HFpEF, LVEF > 50%), HF with reduced LVEF (HFrEF, LVEF < 40%).[11]This review of basic and additional mechanisms of HF progression will contribute to a more comprehensive knowledge of the disease pathogenesis.
NERVOUS SYSTEM ACTIVATION IN HEART FAILURE PROGRESSION
The activation of the systemic and cardiac SNS is the fastest adaptive response mechanism in HF.[12]The SNS activation has some negative effects as well: the release of catecholamines can potentiate different arrhythmias and may aggravate myocardial ischemia. Furthermore, plasma epinephrines are also well-known to have direct toxic effect on cardiac myocytes and induce their hypertrophy and apoptosis. Moreover, norepinephrine causes different signal-transduction abnormalities, such as downregulation of β1- or uncoupling of β2-adrenergic re-ceptors.[12]The activation of β1-receptors can result not only in reflectory tachycardia but also in malignant ventricular arrhythmias. The maladaptive systemic and regional vasoconstriction further leads to different organ failures, such as pulmonary hypertension, renal failure.[13]
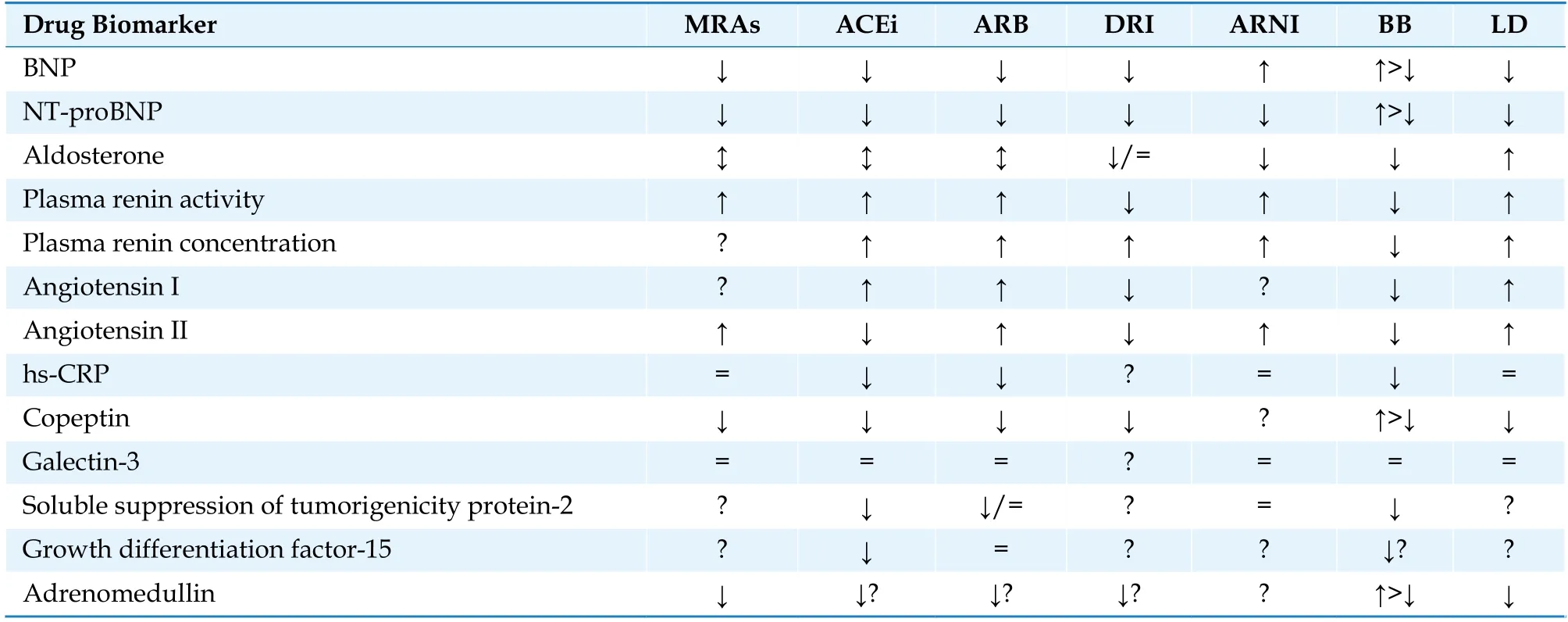
Table 1 Drug regimens for heart failure and their effect on various circulating biomarkers.
Activation of the SNS and inhibition of the parasympathetic nervous system represent one of the first maladaptive mechanisms in disease onset and progression. These processes also form the neuroendocrine model of HF, given its vasoconstrictive,profibrotic, and arrhythmogenic effects.[12]The decreased activity of the parasympathetic nervous system results in abnormal autonomic modulation and reduced heart rate variability.[14]
Circulating norepinephrine increases with disease severity. Data from the Val-HeFT have shown that treatment with valsartan could blunt the increase in norepinephrine compared to placebo,while it was not affected by the mineralocorticoid receptor antagonist spironolactone.[15]Together with catecholamines, chromogranin-A is a component of chromaffin granules in the adrenal glands and, although its biological effects on the cardiovascular system remain to be elucidated, it seems to be involved in the regulation of adrenergic system.[15-17]Limited evidence exists on circulating chromogranin-A and it’s increase in either acute or chronic HF with possible prognostic value.[17]
In HF, sympathetic signaling pathways are activated, cardiac beta receptor number, density, and activity are reduced with decreased catecholamine sensitivity, Gsα and adenylyl cyclase become downregulated. These are all rate limiting steps in the signaling pathway.[18]Such alterations can be interpreted as compensatory protecting effects, which preserve the heart from arrhythmias, apoptosis, and cardiac hypertrophy. However, the same alterations may also lead to functional deterioration through energy starvation. Further contributing factors are the G-protein coupled receptor kinases(GRKs), which are significantly upregulated and activated in HF progression through the heart beta receptor signaling pathways.[19]
Beta-adrenergic signaling pathways play a pivotal role in HF progression.[18]Cardiomyocytes express all β-adrenergic receptor subtypes. Harmful effects of the misbalance in cardiac adrenergic receptors are listed in Table 2.[12,13,16,18,19]
Along with norepinephrine, other neuromakers have been evaluated in association with the pathogenesis, severity and further progression of HF.
Neuropeptide Y (NPY) is a relatively long halflife sympathetic co-transmitter. It is mainly distributed in central and peripheral nervous system. NPY and catecholamines are released from cardiac sympathetic nerves that supply vasculature, cardiomyocytes.[20]NPY augments effects of angiotensin II,leads to vasoconstriction, increases cardiomyocyte calcium loading, participates in cardiac remodeling and potentiates angiogenesis.[21]In patients with congestive HF elevated levels of plasma NPY has been reported years ago.[22]Recently, in an intervention study NPY was measured in coronary sinus blood samples in patients with stable congestive HF. During the mean follow-up period of 28 months the higher adjusted levels of NPY were associated with more adverse events, including death,left ventricle assist device implantation and heart transplant (HR = 9.5, 95%CI: 2.92−30.5, P < 0.001).[23]
Catestatin is a bioactive substance, one of the cleavage products of the pro-hormone chromogranin A. This is a pleotropic peptide secreted together with catecholamines. Catestatin can function as an autocrine negative feedback-loop that limits further catecholamine secretion.[24]Catestatin has been found to play a role in the regulation of blood pressure, cardiac contraction and relaxation, in promotingangiogenesis, and regulating immunity.[25]In one study the progression of HF was associated with a gradual decrease in plasma Catestatin level.[26]Interestingly, in a more recently published cross-sectional study, Catestatin level was increased in patients with acutely decompensated HF of ischemic etiology, suggesting that it is a marker of increased or residual sympathetic activation, which might help to identify HF patients with advanced disease burden and/or an increased risk of post-discharge mortality.[21]

Table 2 Role of cardiac adrenergic receptors activation in heart failure progression.
CIRCULATION BIOMARKERS OF HEART FAILURE PROGRESSION
One of the first systems to be activated in response to impaired cardiac function are RAAS and vasoactive peptides.[26]Humoral activation leads to transcriptional and posttranscriptional changes in the genome, especially in the genes regulating the structure and mechanics of cardiomyocytes. There are also several biomarkers involved in the maladaptive processes in HF progression (Table 3).[26-30]
Natriuretic Peptides
Human brain natriuretic peptide (BNP) and the amino-terminal fragment of proBNP (NT-proBNP)are produced in equimolar fashion from the cleavage of their 108-amino acid precursor proBNP by proprotein convertases, such as corin and furin.[31,32]The biologically active BNP is rapidly degradedin vivoby several peptidases, such as dipeptidyl peptidase IV and neutral endopeptidases (NEP,neprilysin).[33]BNP, together with NT-proBNP, is mainly a product of ventricular myocytes in response to increased myocardial wall stress due to volume or pressure overload. It plays a major role in HF progression, given its diuretic, natriuretic,vasodilator, and anti-hypertrophic properties.[33,34]
Since the beginning of the 21stcentury, several studies have shown clinical utility of B-type natriur-etic peptide testing, supporting the use of this circulating biomarker to diagnose and assess severity and progression of HF.[33-35]At present, BNP and NT-proBNP are routinely used for HF management in a large variety of clinical settings. Strong evidence exists supporting the use of natriuretic peptide testing to diagnose HF in patients presenting with dyspnea. The Breathing Not Properly and the ProBNP Investigation of Dyspnea (PRIDE) trials have shown that BNP and NT-proBNP can diagnose HF in patients with shortness of breath admitted to the emergency room with high accuracy and a significant negative predictive value for levels of BNP < 100 ng/L.[36]Although with lower cut points, the diagnostic value of natriuretic peptides has been confirmed in ambulatory settings.[37]Moreover, in a cohort of around 800 patients with chronic HF, natriuretic peptides have been shown to be associated with the presence of LV systolic dysfunction with an AUC of 0.803 (95% CI: 0.757−0.849) and 0.730 (0.681−0.778) for BNP and NTproBNP, respectively.[38]In stable patients with HFrEF, BNP circulating levels reflect disease severity, and increase with worsening symptoms (NYHA class).[39]The assay of natriuretic peptides is currently recommended for diagnostic purposes by all major scientific societies, including the American College of Cardiology, the American Heart Association, the Heart Failure Society of America, and the European Society of Cardiology.[11]However, there is no such thing as diagnostic cutoff, which often puts natriuretic peptide into a “grey zone.” Further,both BNP and NT-proBNP are influenced by gender, age, and comorbidities, in particular by renal function. Therefore, their interpretation must take into account the overall clinical assessment.[40]

Table 3 Circulating biomarkers of heart failure progression.
Most studies have investigated the prognostic role of admission natriuretic peptide levels.[41,42,43]However, there is evidence that discharge BNP and NT-proBNP levels, and how they change during hospitalization, also predict outcome of patients with HFrEF. Data from the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF) showed that the addition of discharge BNP testing to a clinical model significantly improved risk classification for 1-year mortality with an NRI of 5.5%,[41]thus suggesting that serial natriuretic peptide testing may be useful in pre-discharge clinical decision making.
B-type natriuretic peptides represent the most widely studied cardiac hormones, both in the experimental and clinical field. Atrial natriuretic peptide(ANP) was first described in early 1980s, as a substance secreted from atrial granules with endocrine functions, by Adolfo de Bold.[42]ANP assay has with time been substituted by assays of B-type natriuretic peptides for both analytical and clinical reasons.[43]More recently, the potential clinical applications of the mid-regional MR-proANP with a longer half-life than ANP, have been tested. Data from the Biomarker in the Acute Heart Failure(BACH) and the PRIDE study have demonstrated a good performance of MR-proANP for diagnostic and prognostic purposes in acute HF, even in addition to NT-proBNP.[36,44,45]MR-proANP also revealed a significant prognostic value in 1 237 patients with chronic HF enrolled in the Gruppo Italiano per lo Studio della Sopravvivenza nell’Insufficienza Cardiaca-Heart Failure (GISSI-HF) study,outperforming other established and candidate biomarkers (including copeptin and mid-regional proadrenomedullin, MR-proADM, and C-terminal proendothelin-1), beyond NT-proBNP and a set of clinical variables.[46]
Thus, natriuretic peptides are the cornerstone biomarkers in HFrEF. Nonetheless, their clinical value has been demonstrated across the whole spectrum of LV systolic function. Both BNP and NT-proBNP are key elements in the diagnosis of HFpEF, and their levels are increased in these patients, but to a lower extent compared to patients with more severe reduction in EF. Moreover, their levels increase with more severe cardiac morphological and functional abnormalities (including hypertrophy,fibrosis, and diastolic dysfunction).[47]
Renin-Angiotensin-Aldosterone System (RAAS)Activation
RAAS is a complex endocrine system regulating kidney, liver, vascular endothelium, and adrenal cortex function, salt/water homeostasis and participating in vasomotion. Either systemic or tissue RAAS are involved in tissue remodeling after damage, and can promote fibrosis, hypertrophy, and apoptosis.[48]
The major bioactive molecule of the RAAS system is angiotensin II with its endocrine, autocrine,paracrine, and intracrine effects. It has various effects throughout the body and induces several pathways to regulate arterial pressure and sodium/water balance. Angiotensin II is a general vasoconstrictor in all arterioles with a marked effect on the renal efferent arterioles. It stimulates the release of aldosterone, induces the excretion of noradrenalin from the sympathetic nerve terminals,[33]and inhibits the vagal tone.[49,50]Angiotensin II is not only a vasoactive hormone, but it also has local positive inotropic, negative lusitropic effect on the heart,and increases the afterload resulting in higher energy production by the heart. Angiotensin II has also a direct effect on cardiomyocytes in HF progression by promoting hypertrophy, myocyte apoptosis, and causing structural and biochemical alterations in the extracellular matrix.[51]
Levels of all RAAS effectors are increased in chronic HF, and RAAS activation is an indirect or direct target of most effective pharmacological treatments in HF progression, such as beta-blockers,inhibitors of angiotensin-converting enzyme, angiotensin receptor blockers, direct renin inhibitors, mineralocorticoid receptor blockers, and angiotensin receptor/neprilysin inhibitors. Circulating biomarkers of RAAS activation with varying accuracy are currently available, such as plasma renin activity, renin, angiotensin II, and aldosterone. Some of these biomarkers are well-recognized prognostic factors,even in patients with optimal therapy. Notably,chronic use of drugs targeting RAAS induces, per se, neurohormonal reactivation.[51]
The above-mentioned data support the rationale for the use of biomarkers of RAAS activation as a guide for predicting HF progression and for timely treatment initiation.
The results of experimental studies have demonstrated that plasma renin activity and active renin concentration are longitudinally increased with HF progression, and in symptomatic HFrEF their levels are the highest.[15,16]Although the prognostic role of renin enzymatic activity as a predictor of presymptomatic and symptomatic HFrEF is disputed, the value of plasma renin activity has been reproducibly reported.[52]
In a prospective study renin activity was independently associated with worse prognosis in patients with HFrEF (n = 996), irrespective of medical treatment and similar to other established markers,NT-proBNP and LFEF.[53]Elevated renin activity levels demonstrated increased risk for congestive HF and a trend towards higher mortality among patients with hypertension.[54]
Angiotensin converting enzyme (ACE) inhibitors generally used in patients with HF, and are known to increase plasma renin activity level. However,their presence did not result in poorer prognostic significance of renin activity in Val-HeFT trial.[55]Active renin concentration was found to be superior to plasma renin activity for the assessment of HF severity and in the prediction of survival in HF hospitalized patients with HFrEF and receiving ACE inhibitors.[56]
Cardiac Troponins
Cardiac troponins are released into the circulation following the disruption of cardiomyocyte membrane due to cardiac injury, namely after cardiac necrosis, and have become the standard of care biomarker for diagnosis of myocardial infarction.Troponin is an intracellular protein essential in the regulation of muscular contraction. It is made up of three subunits, Troponin I, T, and C. Cardiac troponins I (cTnI) and T (cTnT) are unique to cardiomyocytes, and unlike Troponin C, they are not expressed in skeletal muscle.[57]Therefore, increased levels of circulating cardiac troponins (cTn) are highly specific for ongoing myocardial damage, and they have been utilized for the past two decades as markers for defining myocardial infarction.[58]It is much less studied that cardiac troponin levels may also be elevated in both acute HF and in decompensation of chronic HF, as well as in other settings with less clear mechanisms of myocardial injury such as septic shock, pulmonary embolism,myocarditis, drug-induced cardiotoxicities, and renal dysfunction.[57-59]Significantly higher cardiac troponin levels observed in patients with more severe infections, patients admitted to intensive care units or in those who have died.
One possible explanation for elevated cardiac troponins in chronic HF may arise from reversible or irreversible myocardial supply and demand mismatch.[59]cTn release may be caused by both acute and chronic myocardial stress, as well as by chronic sub-clinical and sub-endocardial ischemia, or directly related to cardiomyocyte injury. This release of troponin may also indicate increased cardiomyocyte turnover in the setting of progressing myocardial dysfunction. This could explain how cardiac troponins may be detectable in non-cardiac situations, where metabolic demand increases substantially relative to supply, such as hypotension and shock. An alternative explanation for higher circulating cardiac troponin levels may be related to diminished renal clearance rather than intensified ongoing myocardial damage.[60]In the general population, elevated cTnT marks subclinical cardiac injury and elevations of cTnT quantified via high sensitivity assays indicate an increased risk for structural heart disease and all-cause mortality.[61]Interestingly, circulating levels measured by newer-generation,highly-sensitive cTn assays are frequently detectable even in non-HF patients. Furthermore, levels of cTn that are detected using these assays are independently associated with all-cause mortality, cardiovascular mortality, and incident HF in the general population, despite controlling for renal function,NT-proBNP, and hs-CRP.[62]
A recent meta-analysis involving 16 studies of cTn levels in patients with chronic systolic or diastolic HF, revealed that cTn levels predict both allcause and cardiovascular mortality and adverse cardiovascular outcomes.[63]These trends persisted despite changes in medical therapy. High sensitivity cTnI and cTnT assays detect levels of cTn much lower than 10 ng/L, which is below the 99th percentile decision limit for assisting in the diagnosis of myocardial infarction. With such cTn levels, there is a decrease in diagnostic specificity when suggesting myocardial infarction. This questions the utility of these assays in an acute coronary syndrome, but also raises the possibility that high sensitivity cTn assays may be used to stratify risk in stable chronic patients. Patients with chronic HF typically have elevated cTn when measured with high sensitivity assays compared to standard cTn assays (detected in 63.9% vs. 31.1% patients).[64]In a study with a very high proportion (92%) of subjects with chronic HF progression and positive high-sensitivity cTn suggested a weaker association of high-sensitivity cTn assays with prognosis when compared to the standard assays. Although, this trend has not been replicated in other studies.[65]To fill this gap, it is important to perform prospective studies determining the prognostic significance of detectable cTn levels measured with high sensitivity assays in patients with chronic HF and undetectable cTn via standard assay.[64]This would, ideally, identify a population at higher risk that may benefit from intensified therapy.
BIOMARKERS OF FIBROSIS
Transforming Growth Factor-β1 (TGF-β1)
TGF-β1 is a powerful fibrogenic cytokine and an important regulatory cytokine involved in tissue repair, whose sustained production in many tissues underlies the development of fibrosis.[66]TGF-β1 expression is increased in almost all fibrotic diseases.There is growing evidence supporting a causative role of oxidative stress in fibrogenesis in various tissues including liver, lung, arteries, nervous system and heart.[66,67]Cardiac fibroblasts are indeed involved in post myocardial infarction remodeling through the generation of replacement scar tissue in the infarct zone and through production of fibrosis in unaffected myocardial segments.[66]TGF-β1 released from infiltrating lymphocytes, platelets, activated macrophages, injured myocytes and fibroblasts, increases the deposition of extracellular matrix at the site of myocardial tissue injury, in order to repair the damage. This occurs through stimulation of new matrix proteins such as collagens, fibronectin and proteoglycans, through inhibition of proteases and stimulation of the synthesis of protease inhibitors.[68]TGF-β1 also increases the expression of cellsurface integrin receptors, so that the cell-matrix interaction and matrix ansembly are enhanced. The biological effects of TGF-β1 are even more amplified by autoinduction of its own production by these cells. TGF-β1 binds to proteoglycans in the matrix or near the cell surface, which may serve as a signal to terminate the production of TGF-β1 after tissue repair is complete.[69]During repeated injury there is a continuous autoinduction of TGF-β1 leading to an overproduction of TGF-β1 and to a continuous production of extracellular matrix and to tissue fibrosis.[68]This overproduction overrides the termination signal and thereby creates a chronic vicious circle of TGF-β1 overproduction.[69]Reactive oxygen species have been shown to upregulate the expression of TGF-β1 and collagen type I in various tissues.[67]Therefore, TGF-β1 seems to have a crucial role in cell proliferation, differentiation, migration and extracellular matrix production in the myocardium.[70]
In various experimental animal models as well as in humans, an enhanced cardiac TGF- β1 expression is associated with appearance of myofibroblasts and activated collagen synthesis, while collagen degradation is suppressed.[71]As TGF-β1 is involved in different cardiac pathological processes, it seems like a sure candidate target for inhibition.Animal studies have shown that inhibition of TGFβ1 mediated effects may attenuate cardiac fibrosis and HF progression. However, the potential side-effects of TGF-β1 inhibition, such as toxicity and immunological alterations, must be weighed carefully before its clinical use.[72]While the beneficial effects of angiotensin converting enzyme inhibition and angiotensin II type I receptor blockers on left ventricle remodeling have been established, adverse left ventricle remodeling still occurs in a substantial proportion of heart failure patients. Novel therapeutic strategies such as activation of cardiac and leucocyte oxidative stress pathways, activation of inflammatory pathways and matrix metalloproteinases and eNOS-derived nitric oxide availability are under investigation for potential application.
Galectin-3 (Gal-3)
Gal-3, a soluble β-galactoside-binding lectin, has been found to play an important mechanistic role in the development of cardiac fibrosis and remodeling and to identify high-risk subsets in HF cohorts, thus qualifying both as a risk marker and a risk factor.[73,74]There is a growing amount of evidence that Gal-3 is essential for migration and phagocytic activity of macrophages. Macrophage-derived Gal-3 may then act on fibroblast proliferation and on collagen synthesis, by increasing collagen I and reversing the collagen I-to-collagen III ratio.[75]Together with liver and kidney fibrosis, Gal-3 has been strictly linked to the development of cardiac fibrosis, being a key determinant of cardiac remodeling and HF progression, possibly by interacting with mechanisms of aldosterone-mediated damage.[76]
The experimental data of a mechanistic involvement of Gal-3 in fibrotic, inflammatory, and remodeling processes in HF progression led to a novel interest in the potential use of Gal-3 level in plasma,as a biomarker. van Kimmenade and colleagues in 2006 first compared NT-proBNP, apelin, and Gal-3 in the management of acute HF patients.[77]Out of 599 acutely dyspneic patients, 209 were later diagnosed with HF. Although Gal-3 showed a limited diagnostic accuracy in identifying acute HF, it was the strongest predictor of early events (60-day rehospitalization for HF or all-cause mortality). Later,Gal-3 was also demonstrated to predict long-term outcome in another cohort of acute HF patients(proposed cutoff: 14.97 ng/mL), independently of echocardiographic indices of cardiac structure and function (LV end-diastolic/systolic diameter, EF and right ventricular pressure).[78]In the following years the prognostic role of Gal-3 in HF settings has been investigated in some substudies from larger clinical trials. Gal-3 levels were determined in 232 NYHA III-IV chronic HF patients enrolled in the Deventer-Alkmaar HF (DEAL-HF) study, who were then followed up for a period of 6.5 years.[79]Baseline Gal-3 (cutoff: 17.6 ng/ml) predicted all-cause mortality after adjustment for age, gender, creatinine clearance, and NT-proBNP. By dichotomizing the population according to NT-proBNP and Gal-3 levels, the authors also demonstrated an additive prognostic power for Gal-3, since patients with elevation of both biomarkers had a 1.5- to 2-fold higher mortality rate compared to patients in other subgroups. In a larger population of 895 chronic HF patients from the HF-ACTION study with LVEF <35% (Heart Failure: A Controlled Trial Investigating Outcomes of Exercise TraiNing), Gal-3 lost its univariate prognostic value in predicting the composite outcome of all-cause death or re-hospitalization,when corrected for peak oxygen consumption at cardiopulmonary test or NT-proBNP.[80]
Despite this wide amount of data confirming its prognostic role in HF, there is still limited information on whether Gal-3 assay may help in adjusting therapeutical strategies. Current knowledge is indeed limited to a benefit from statin therapy in patients with chronic HF of ischemic cause and low Gal-3 level,[81]and to the lack of power in predicting response to mineralocorticoid receptor antagonists or cardiac resynchronization therapy.[81,82]
Recently, measurement of Gal-3 has received a class IIb recommendation in acute decompensated and in chronic HF for risk stratification purposes in the 2013 ACCF/AHA Guideline for the Management of Heart Failure.[83]
Gal-3 has also been mechanistically involved in the processes of cardiovascular inflammation and fibroblast proliferation and fibrosis, which are thought to be involved in the development of HFpEF and HF decompensation. In 2011 de Boer has shown that in HFpEF patients from the Coordinating study evaluating Outcomes of Advising and Counseling in Heart Failure (COACH) (with LVEF >40%), Gal-3 appeared to have a particularly strong predictive value, compared to HFrEF patients.[73]Gal-3 also yielded significant reclassification indices in one of the largest biomarker studies in HFpEF so far, conducted in a cohort of 419 HF patients with LVEF > 45%.[84]The Aldo-DHF trial explored the effects of spironolactone 25 mg vs.placebo in chronic HFpEF patients. Gal-3 was associated with functional performance and quality of life, and its increase in serial measurements predicted all-cause death or hospitalization independently from NT-proBNP. However, no specific interaction between treatment arm and Gal-3 levels could be observed.[85]
In a canine model of HFpEF induced by aortic banding, myocardial galectin-3 was significantly upregulated after two weeks. Increase in galectin-3 expression positively correlated to the severity of diastolic dysfunction assessed with the echocardiographic diastolic parameter—early transmitral flow velocity to early diastolic tissue velocity (E/Em) ratio.[86]Although it is evident that galectin-3 plays a crucial role in cardiac fibrosis and in HF, some studies have demonstrated the significance of galectin-3 in maintaining the integrity of cardiac tissue after necrosis. In a mouse model of myocardial infarction, galectin-3 KO displayed an increased trend towards mortality, chiefly due to ventricular rupture,[87]emphasizing that galectin-3 is necessary for normal wound healing, especially during the initial phases of cardiac repair.
Studies in mice deficient for galectin-3 have suggested that galectin-3 is not a bystander but rather a culprit for myocardial fibrosis. Inhibition of galectin-3, either achieved with large carbohydrates, or antisense RNA, or small designer molecules, effectively reduces organ fibrosis.[88]
Suppression of Tumorigenicity Protein-2 (ST2)
ST2 is a member of the Toll-like/ interleukin-1 receptor superfamily, which is expressed together with its ligand interleukin-33 following myocardial stretch and cardiovascular injury. The interleukin-33/ST2 interaction exerts positive effects in terms of blunting prohypertrophic and profibrotic signals.[89]Indeed, the transmembrane receptor for interleukin-33 - ST2 ligand (ST2L) is one of ST2 isoforms. Among others, a soluble isoform of ST2 (sST2) exists, arising from a dual-promoter system driving differential mRNA expression. There is evidence that sST2 may act as a decoy receptor competing with ST2L for interleukin-33 binding. For example, administration of interleukin-33 to cultured rat neonatal cardiomyocytes inhibited the prohypertrophic signals of angiotensin II or phenylephrine, these effects being reversed by sST2.[90]Reflecting these experimental evidences, serum levels of sST2 correlated with left ventricle systolic function and remodeling, and were associated with more decompensated hemodynamic profile.[91]Notably, sST2 concentration does not appear to be influenced by age, kidney function,or body mass index, unlike natriuretic peptides.[92]
Among markers of inflammation and fibrosis,sST2 seems to be promising in the clinical management of HFpEF. First data came from a prognostic study showing that sST2, although not correlating with echocardiographic indices of diastolic function, was the only humoral marker predicting mortality in 200 patients with dyspnea and normal left ventricle systolic function.[93]These data have been confirmed in further studies, demonstrating that prognostic value of sST2 in HFpEF was comparable to that in HFrEF, especially in acute settings.[94]
sST2 assay has been tested for diagnostic and prognostic purposes in either acute or chronic HF populations. Data from the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE)study have shown higher sST2 concentration in subjects with acute HF compared to those with dyspnea of non-cardiac origin.[93]In the same cohort,sST2 was reported to be the best predictor of 1-year death, outperforming NT-proBNP.[95]Interestingly,in a population of acute HF patients, most of them with left ventricle systolic dysfunction, sST2 provided additional prognostic value over NTproBNP in the prediction of death, even in subset with lower NT-proBNP.[96]Fewer studies have explored the prognostic value of sST2 in chronic HF decompensation. In a well-treated subset of patients with LVEF < 35% enrolled in the HF-ACTION trial, sST2 was only modestly associated with functional capacity while it was significantly associated with outcomes.[97]
After initial adjustments for conventional variables in CORONA study, baseline sST2 was a significant predictor of all endpoints, including the primary endpoint, death, worsening HF, and hospitalization for HF. When NT-proBNP and C-reactive protein were added to the model, sST2 was no longer a predictor of primary outcome, but remained significantly predictive of death from worsening HF, cardiovascular hospitalization, and hospitalization for HF worsening.[98,99]
In another study, sST2 value was also available at 4 and 12 months and patients randomized to the valsartan arm showed a milder increase in sST2 over time compared to those assigned to placebo.Further, in an analysis including also growth differentiation factor-15 (GDF-15) and high sensitive troponin-T, sST2 was the only circulating biomarker to add prognostic information to baseline concentration and to predict the occurrence of reverse remodeling when serially tested in decompensation chronic systolic HF patients.[100]Based on these data, and on the association of neurohurmonal antagonism therapy with lower sST2 concentrations, sST2 is particularly promising as a tool for patient monitoring and therapeutic optimization.[95]Further, as for Gal-3, sST2 measurement has received a class IIb recommendation in acute decompensated (level of evidence A) and in decompensation of chronic(level of evidence B) HF for risk stratification purposes in the latest American guidelines.[83]
BIOMARKERS OF INFLAMMATION
There is growing evidence on the role of inflammatory cells and pathways during acute cardiovascular injury and in the reparative process that is subsequently activated. Elevation of inflammatory biomarkers, including C-reactive protein (CRP),members of the interleukin family (e.g., IL-1, IL-6,and IL-18), and TNF-α, is a hallmark feature of chronic ischemic and non-ischemic HF. It remains debatable whether inflammation is causative to disease progression.[101]Moreover, viral infection is thought to participate in the development of dilated cardiomyopathy, sustaining acute and chronic inflammation.[102]
The Val-HeFT study demonstrated a direct correlation between elevated CRP level and HF severity.Further, CRP predicted the risk of death and early readmission in acutely decompensated HF.[103]Antiinflammatory properties have been described for statins and their effect has been tested in HF settings. Data from the CORONA study showed that subjects with HF of ischemic etiology and elevated baseline hs-CRP (2 mg/L) exhibited a greater benefit from statin therapy in terms of reduction of the primary end-point of cardiovascular death, myocardial infarction, and stroke,[99]while controversial results come from studies performed on patients with HF of non-ischemic origin.[104]
Some trials have addressed TNF-α elevation in HF progression, which is associated with worsened prognosis. Two large clinical studies, the Research into Etanercept Cytokine Antagonism in Ventricular Dysfunction (RECOVER) and the Randomized Etanercept North American Strategy to Study Antagonism of Cytokines (RENAISSANCE), were stopped because of lack of clinical benefit, and patients receiving the highest dose had increased adverse cardiac outcomes.[105]Similar results were observed in the Anti-TNF-α in Congestive Heart Failure (ATTACH) trial, testing humanized neutralizing antibodies (infliximab) instead of the soluble receptor etanercept.[106]The negative results of such trials may be, at least in part, explained by the inappropriate blockade of physiological inflammation that may be required for tissue-reparative processes.
OTHER BIOMARKERS
Copeptin
Arginine vasopressin (AVP), also known as antidiuretic hormone (ADH), is involved in the regulation of free water clearance, plasma osmolality, and vasomotricity. It has been known for a long time that circulating AVP is elevated in HF settings,[107]but its detection is challenging. Copeptin, the c-terminal segment of the precursor of provasopressin,is a reliable surrogate marker for AVP, which has been proved to independently predict mortality in acute HF, especially in subsets with hyponatremia.[108]In 195 patients with chronic HFrEF, copeptin predicted 5-year all-cause mortality, although it’s additive prognostic accuracy over NT-proBNP was poor.[109]
Adrenomedullin (ADM)
ADM is a peptide hormone acting as a potent vasodilator and is expressed by all human tissues.Circulating ADM is increased in HF and correlates with disease severity. Nonetheless, ADM is highly unstable and, recently, a novel assay measuring the mid-region of the more stable prohormone (MRproADM) with similar behavior in HF patients has been developed.[110]Prognostic value of ADM has been tested in a cohort of 297 patients with HFrEF of ischemic origin. In this study ADM predicted the risk of mortality and of HF hospitalization independently from other clinical variables.[111]Since elevation of ADM is a feedback response to volume overload to maintain vascular integrity and decrease vascular leakage of fluid from the the vasculature to the tissues, measurement of ADM might reflect tissue and pulmonary oedema. Such a measurement might guide physicians to more intensively treat patients with heart failure hospitalization and facilitate discharge decisions. In addition,ADM might become a target of therapy in heart failure.[110]
Chemokines
Chemokines are chemotactic cytokines, small glycoproteins, which stimulate leukocyte migration,regulate cytokine production, and may induce reactive oxygen species (ROS) formation as a response to an acute inflammatory event. There are four known chemokine subfamilies, differentiated according to the primary amino acid sequences:CXC, CC, C, and CX3C. The plasma concentration of certain chemokines, such as MCP-1α and RANTES increases significantly in HF.[112]There are also neutrophil-specific chemokines, the CXC chemokines,including IL-8, neutrophil activating peptide-2 and GRO-α, whose elevated level is proportional to the severity of the initiating disease. For instance, overexpression of myocardial CXCR4 has been shown to result in enhanced recruitment of inflammatory cells, increase of TNFα production, accelerated apoptosis and cell turnover in animal model.[113]Furthermore, overexpressed CXCR4 in mesenchymal stem cells induced effectively neomyoangiogenesis in the infarcted myocardium.[113]
Fibroblast growth factor (FGF)
Serum levels of FGFs have been associated with established cardiovascular risk factors as well as with the severity and extent of coronary artery disease and could be useful for prediction of cardiovascular death. Furthermore, preclinical investigations and clinical trials have tested FGF administration for therapeutic angiogenesis in ischemic vascular disease, demonstrating a potential role in improving angina and limb function. FGF21 has lately emerged as a potent metabolic regulator with multiple effects that ultimately improve the lipoprotein profile. Early studies show that FGF21 is associated with the presence of atherosclerosis and may play a protective role against plaque formation by improving endothelial function. The present review highlights recent investigations suggesting that FGFs, in particular FGF21, may be useful as markers of cardiovascular risk and may also serve as protective/therapeutic agents in cardiovascular disease.[114]
Serum FGF21 is elevated in multiple models of HF, and appears to have both cardiac and extra cardiac sources. Future work will investigate 1) whether there is a correlation between FGF21 levels and mitochondrial damage, and 2) the signaling pathway resulting in metabolic stress to other organs in HF.[115]
Adipokines
Сardiovascular effects of adipokines include direct actions on target tissues and effects occurring as a result of central stimulation of the sympathetic nervous system.[116]Adipokines play a regulatory role in myocardial function through their involvement in myocardial metabolism, myocyte hypertrophy, cell death, and changes in the structure and composition of the extracellular matrix.[117]The direct regulation of myocardial remodelling components (matrix metalloproteins, tissue inhibitor of metalloproteinases and collagens) by adipokines has been demonstrated in both in vitro and in vivo studies.[116-118]
Adiponectin and leptin have been implicated in heart disease and myocardial remodeling.[119]Adiponectin levels are reduced in obese people while leptin levels are positively related with BMI and adiposity. Adiponectin regulates cardiac injury by modulating antiinflammatory and prosurvival reactions, and inhibiting cardiac remodeling.[120]It has also been reported that decreased levels of adiponectin are associated with hypertension through various mechanisms including the renin angiotensin system and sympathetic nervous system hyperactivity, endothelial dysfunction and renal pressure natriuresis impairment.[121]
Hypoadiponectinaemia appears to exacerbate aortic stiffness in primary hypertension subjects.[119]Therefore, one would expect hypoadiponectinaemia to be an important link to obesity-related hypertension; however, it appears that hypoadiponectinaemia results in hypertension in lean subjects,independently of risk factors.[122,123]
The effects of adiponectin are mediated via its potentiation of insulin action, increase in NO production, stimulation of prostaglandin synthesis via inducible cycloxygenase-2, and activation of adenosine monophosphate, which also activates protein kinase and inhibits α-adrenergic-receptors stimulating hypertrophy of cardiac myocytes.[124-126]Adiponectin modulates tissue ceramide levels by activating ceramidase which hydrolyses ceramide.[127]Animal studies have suggested that adiponectin deficiency is linked to myocardial damage, HF and cardiac hypertrophy.[123,128,129]
However, leptin exerts cardioprotective effects directly against ischaemic-reperfusion injury.[130]Leptin stimulates cardiac hypertrophy directly through complex cell signalling mechanisms and indirectly through its effects on hypertension and the sympathetic nervous system and indirectly by producing a signal which limits cardiac lipotoxicity.[131,132]
Leptin is a negative inotrope (mediated by NO)and it exerts its effects primarily in cardiomyocytes acting via binding to its receptors and subsequent activation of various kinases in cardiomyocytes.[133]Leptin stimulates cardiac hypertrophy directly through complex cell signaling mechanisms, and indirectly through its effects on hypertension and the sympathetic nervous system, as well as by producing a signal which limits cardiac lipotoxicity.[134]Leptin regulates cardiac contractility, metabolism,cell size and production of extracellular matrix substances in cardiomyocytes.[135]Leptin increases oxidative stress in endothelial cell, promotes smooth muscle cell proliferation and migration and vascular calcification. The link between fat mass and atherogenesis has been confirmed by the findings in the obese, leptin-deficient mouse as described earlier.A second proposition for the link between vascular dysfunction and obesity lies in the fact that CRP levels are elevated in the obese and correlate with increased risk of CVD via inflammation. Because leptin and adiponectin appear to have opposite effects, it is speculated that the leptin/adiponectin ratio may play a role in myocardial remodeling and HF progression.[134,136]
Resistin, an adipokine involved in inflammation and insulin resistance has also been associated with the development of HF after accounting for obesity,insulin resistance, and coronary heart disease.[137]The precise role of resistin in cardiac pathology and ischaemic-reperfusion injury is yet to be defined, although, resistin has been linked to insulin resistance and dyslipidemia, and shown to inhibit vesicular transport of glucose.[137,138]Resistin, like leptin,does not lend itself especially well to benefiting CVD. High levels are associated with insulin resistance, increased cardiovascular risk, unstable angina, endothelial dysfunction, increased proatherogenic inflammatory marker, vascular smooth muscle cell proliferation and poor prognosis in coronary artery disease.[138]
Another adipokine—apelin is a peptide produced and secreted not only by adipocytes, but also by stromal vascular fraction and cardiovascular tissues.[139]Apelin increases the serum adiponectin level and decreases that of leptin. Apelin plays a role regulating insulin resistance by influencing serum adiponectin levels, energy expenditure and expression of uncoupling proteins in brown adipose tissue in mice.[139]Plasma levels of apelin are reduced in subjects with dyslipidemia,[140]and therapeutic reduction in the atherogenic LDL (via statins or lifestyle changes) in patients with isolated hypercholesterolaemia leads to a rise in circulating apelin.[139]In hyperlipidaemic animal models, apelin appears to antagonise angiotensin II-enhanced atherogenesis.[139]It has also been reported that apelin prevents aortic aneurysm formation in a murine model by limiting macrophage inflammation - possibly via chemokine activation and inhibition of inflammatory cytokines.[140]
So, according to numerous data, it is known that decreased plasma levels of apelin have been observed in patients with lone atrial fibrillation and chronic HF in which cardiac resynchronisation therapy helps to improve them.[139,141,142]Animal models of HF demonstrate cardiac apelin to be downregulated by angiotensin II. This cardiac apelin is restored by treatment with angiotensin type 1 blocker.[139]However, myocardial expression of apelin is increased in ischaemic HF. So myocardial expression of apelin may be decreased or increased but plasma levels of apelin are decreased in patients with chronic HF.[139,141]Contributing to the complexity of the picture, myocardial unloading in HF patients (using a left ventricular assist device)results in upregulation of left ventricular apelin expression.[141]Beyond these observations and hypotheses, the cardiovascular role of apelin is not entirely understood and further study is required to clarify this. The cardiac effects of other adipokines such as visfatin, vastin, omentin and chemerin are much less studied.
MicroRNAs (miRNAs)
miRNAs are non-coding RNAs which are involved in different cell processes by repressing messenger RNA translation. miRNAs have been shown to participate in several pathophysiological processes related to coronary artery disease and heart failure, including cardiac fibrosis and hypertrophy.[143,144]Several circulating miRNAs have been tested for diagnostic and prognostic purposes either in acute or in chronic HF.[145]In a small cohort of patients with chronic HF, miR-126 and miR-508-5p were associated with cardiovascular death in patients with ischemic and non-ischemic HF, respectively.[146]Interestingly, there is experimental evidence that circulating miRNAs fluctuate in response to pharmacological HF treatment (e.g., by ACE inhibitors),and that miRNAs may represent, themselves, a therapeutical target.[147]
In chronic HF, multiple miRNAs have been described as candidates for future diagnostic biomarkers in HF progression.[148-151]A few studies have reported on circulating miRNAs that were able to distinguish patients with shortness of breath due to HF and other causes of dyspnea. For example, miR-423-5p had different expression levels in HF patients,healthy controls, and patients with dyspnea of noncardiac origin.[152]MiR-423-5p has been shown to be differentially expressed in HF patients and healthy controls, and patients with other causes of dyspnea.Other studies also described differences in expression of circulating miRNAs in acute HF, including low levels of miR-103, miR-142-3p, miR-30b, and miR-342-3p,21 and high levels of miR-499.22 A recent study by our group identified a panel of acute HF-specific miRNAs.[153]We observed decreased miRNA levels in acute HF patients compared to healthy controls and patients with an acute exacerbation of chronic obstructive pulmonary disease.
In plasma of patients diagnosed with hypertrophic cardiomyopathy without HF symptoms, miR-29a,among others, was found to be significantly upregulated, and the only miRNA to correlate with both LV hypertrophy and fibrosis.[154]Recent evidence suggests miRNAs to help differentiate between a HFrEF and HFpEF.[150−153]
Such role of miRNAs might not only be relevant for diagnostic purposes, but could provide a better insight in differential pathophysiology of HFrEF and HFpEF as well.
EMERGING APPROACHES AND FUTURE DIRECTIONS
Despite recent advances, HF still remains a syndrome with unacceptable morbidity and mortality,possibly reflecting a complex pathophysiology and an extremely heterogeneous presentation which is only partly counterbalanced by a universal therapeutic strategy. Novel biomarkers addressing specific disease phenotypes and paths of damage are of urgent need for the treatment tailoring and for the transition to a more rational use of drugs and devices in HF progression.
For prognostic purposes, it seems reasonable that the use of multiple markers reflecting the activation of different pathophysiological pathways may more accurately identify high-risk subjects.[155]For example, increased levels of at least three factors among hs-TnT, MR-proADM, combined free light chains, hs-CRP, and sST2 provided incremental prognostic value when added to a multivariable model including BNP. Nonetheless, the feasibility and the cost-effectiveness of multimarker strategies remain to be elucidated.
During the last years, a growing number of biomarkers have been proposed as potentially useful in HF patients, but not one of them still resembles the characteristics of the “ideal biomarker.” A single marker will hardly perform well for screening, diagnostic, prognostic, and therapeutic management purposes. Moreover, the pathophysiological and clinical significance of biomarkers may depend on the presentation, stage, and severity of the disease(e.g., on etiology, presence/extent of LV systolic dysfunction, comorbidities). One could envisage specific sets of biomarker with different performances in HFpEF and HFrEF, especially as concerns prediction of the future course of the disease and of LV adverse/reverse remodeling.
Metabolomics is a comprehensive analysis of small molecules (metabolites) which account up to 6 500 in human body.[156]Metabolomics might become a powerful diagnostic tool profiling specific characteristics of particular patient cohorts and individuals with different disease stages and prognosis. In cardiovascular diseases metabolite correlates have been identified in hyperlipidemias, diabetes, and coronary artery disease.[157]
Specific metabolic molecules have been identified associated with incident HF.[158,159]However,the role of metabolomics in HF progression prediction and evaluation needs to be elucidated.
We believe that a wide range of biochemical,neuromarker, instrumental and clinical data of HF patients, signs and symptoms of co-morbidities,data from implantable sensors and cardiac devices will be used in artificial intelligence algorithms for diagnosis, prognosis and therapy decision-making(Figure 1).[160]This approach is being under active investigation by research groups over the world.
CONCLUSIONS
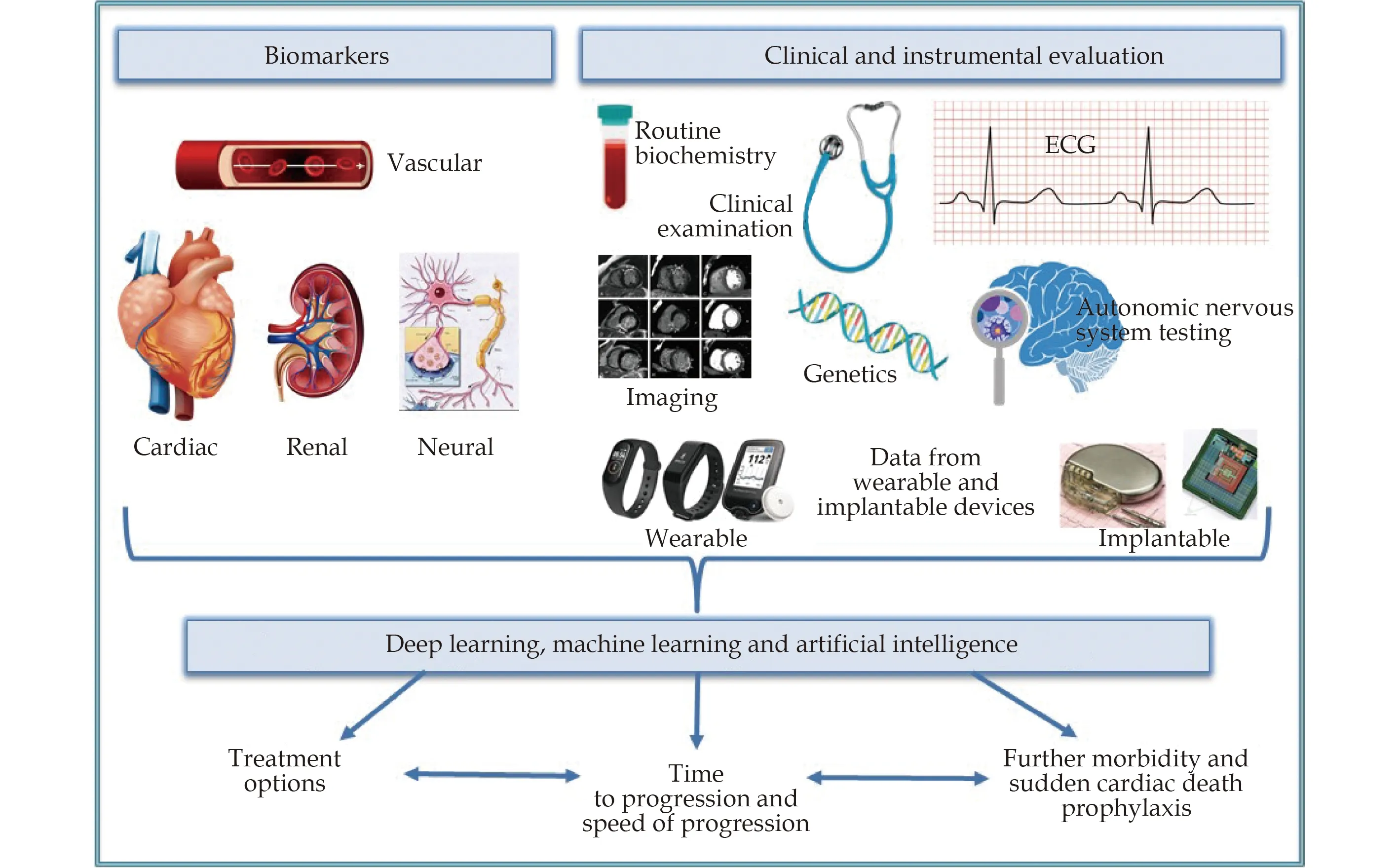
Figure 1 A suggested complex approach to heart failure patients.
Along with established and widely used biomarkers of HF characterization, such as NT-proBNP and hs-CRP, novel substances are extensively being studied. Existing fundamental and pre-clinical evidence proves biomarker evaluation to be a very promising tool in characterization of HF and in personalizing therapy. Despite more than 30 years of clinical research in HF and the approval of many effective therapies for patients with HF, rates of cardiovascular events including hospitalizations,emergency department and office visits, the need for acute interventions and even death remain inadmissible high. Understanding the molecular pathophysiology of HF might serve as a key to identifying new pharmacologic targets for this disease.Clearly, additional innovative and more effective therapies that target new pathways are needed in all patients with HF.
Now it all comes to the quality of clinical studies(even more than results) aimed at improving current diagnostic and prediction models and of studies using biomarkers to tailor therapeutics.
ACKNOWLEDGEMENTS
The study has been supported by t he grant from the Ministry of Science and Higher Education of the Russian Federation (agreement 075-15-2020-800).The authors declare no conflict of interests.
杂志排行

Journal of Geriatric Cardiology的其它文章
- How to do when PFO closure failed under routine guidance:the first clinical experience for PFO closure
- Influence of normal to high stroke volume on congestive heart failure development after transcatheter aortic valve implantation: case series
- Hypertension and its physio-psychosocial risks factors in elderly people: a cross-sectional study in north-eastern region of Bangladesh
- Patent foramen ovale closure in non-lacunar cryptogenic ischemic stroke: where are we now?
- Sex modification of the association of the radial augmentation index and incident hypertension in a Chinese communitybased population
- Obstructive sleep apnea increases heart rhythm disorders and worsens subsequent outcomes in elderly patients with subacute myocardial infarction